Farmacocinética: procesos y evolución de los fármacos en el organismo
Documento de Unie* Universidad sobre farmacocinética. El Pdf explora los procesos que regulan la evolución temporal de las concentraciones de un fármaco en el organismo, incluyendo la distribución y las barreras especiales, para la asignatura de Biología a nivel universitario.
Ver más13 páginas


Visualiza gratis el PDF completo
Regístrate para acceder al documento completo y transformarlo con la IA.
Vista previa
Farmacocinética y Procesos Generales
unie
Universidad
Tema 2. Farmacocinética.
Farmacología I. Bases farmacológicas,
nutrición y dietética.
Grado de Enfermería.
Curso 2024/2025.
Planeta Formación y Universidadesunie
Universidad
Ciclos Generales de la Farmacocinética
La farmacocinética describe el conjunto de procesos que caracterizan la evolución
temporal de las concentraciones de un fármaco administrado, por una vía de
administración específica y en determinadas condiciones. Esos procesos son liberación,
absorción, distribución, metabolismo y excreción.
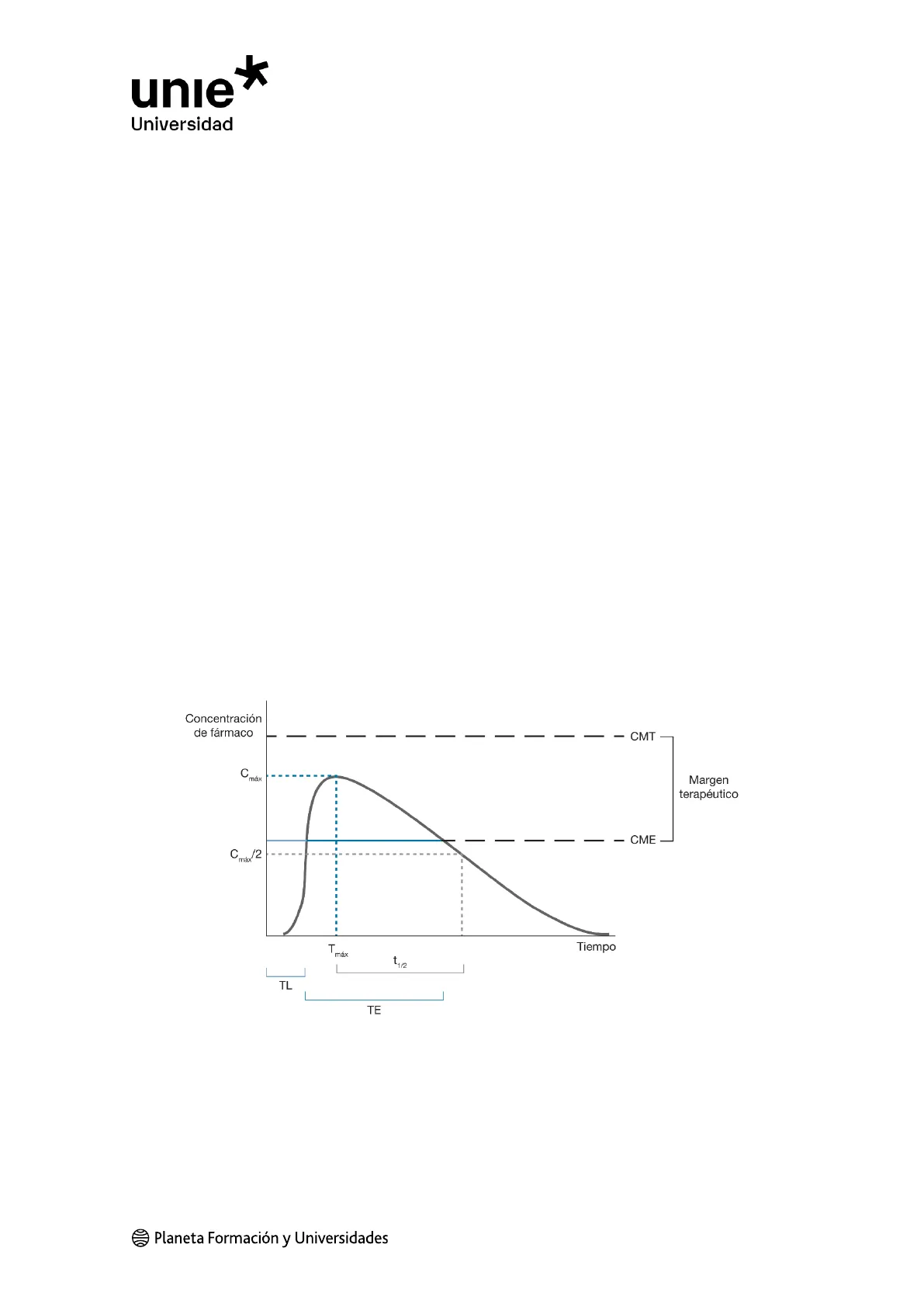
La respuesta terapéutica de los fármacos requiere de una concentración mínima en el
órgano diana (lugar en el que el fármaco es capaz de interactuar con su diana
farmacológica), o concentración mínima eficaz (CME), por debajo de la cual no se
observarán efectos terapéuticos.
Por el contrario, si se alcanzan concentraciones muy elevadas que superen la
concentración mínima tóxica (CMT), se producirán efectos tóxicos. El intervalo de
concentraciones entre la CME y la CMT es lo que se denomina margen o rango
terapéutico.
Por ello, la duración del efecto de un fármaco dependerá del tiempo en el que sus
concentraciones se encuentren dentro de este rango, y la intensidad será directamente
proporcional a la concentración que se alcance.
Concentración
de fármaco
- CMT
C
máx
Margen
terapéutico
- CME
Tiempo
T
máx
t1/2
TL
TE
El tiempo necesario para que se reduzca a la mitad la concentración plasmática,
alcanzada por una dosis de un fármaco administrado, se conoce como semivida de
eliminación (t 1/2 ). El valor de la t 1/2 es uno de los parámetros farmacocinéticos más
importantes, ya que su valor nos indica el tiempo necesario entre la administración de
sucesivas dosis.
Planeta Formación y Universidades
2unie
Universidad
Mecanismos de Transporte a Través de Membrana
Lugar de administración
Liberación
de la forma
farmacéutica
Fármaco
Absorción
Metabolismo local
Metabolismo hepático
Sangre
Fármaco
metabolito
Distribución
Distribución
Tejido 1
Hígado
Fármaco
metabolito
Metabolismo local
Distribución
Distribución
Fármaco
metabolito
Distribución
Fármaco
metabolito
Tejido 2
Excreción
Riñón
Fármaco
metabolito
Orina
Tejido diana
Efecto farmacológico
Para que el fármaco alcance el órgano diana, debe atravesar múltiples membranas; de
la misma forma, para su eliminación debe alcanzar la orina, vía de excreción mayoritaria
para los fármacos, o la bilis. Todos estos procesos implican el paso a través de las
membranas biológicas, de tal forma que estas constituyen una barrera.
- Mecanismos de transporte a través de membrana: El mecanismo general de los
fármacos para atravesar la membrana celular es la disolución en el componente
lipídico.
Procesos Pasivos de Transporte
- Procesos pasivos:
- Se producen a favor de un gradiente de concentración.
- No requieren gasto de energía.
- Filtración a través de poros:
o El fármaco atraviesa la membrana a través de los poros o canales
acuosos, cuyo número y tamaño dependen del tipo de membrana.
Medicamento
Planeta Formación y Universidades
3unie
Universidad
o Solo es aplicable a moléculas pequeñas o las poco ionizadas son capaces
de hacerlo así.
o Este proceso para fármacos por vía intramuscular o subcutánea.
- Difusión pasiva:
o Es el mecanismo más utilizado por los fármacos.
o Ley de Fick: la cantidad de fármaco que atraviesa la membrana y la
velocidad a la que lo hace será tanto mayor cuanto:
· Mayor sea el gradiente de concentración.
· Menor sea el tamaño.
· Mayor sea su liposolubilidad.
Procesos Mediados por Proteínas Transportadoras
- Procesos mediados por proteínas transportadoras:
- Son saturables y muy selectivos.
- Raramente utilizados por los fármacos.
- Difusión facilitada:
o A favor de un gradiente de concentración.
o Este mecanismo es utilizado por moléculas como glúcidos.
- Transporte activo:
o Se realiza mediante el uso de bombas en contra de un gradiente
electroquímico.
o Bomba de sodio/potasio: traspaso de iones de sodio y potasio por la
membrana plasmática. En este caso se requiere energía en forma de
adenosina trifosfato (ATP).
Otros Sistemas de Transporte
- Otros sistemas de transporte:
- Endocitosis y exocitosis: ambos mecanismos son utilizados por
macromoléculas fisiológicas - Liposomas: vesículas esféricas de pequeño tamaño formadas por una
doble capa lipídica que en su interior llevan el fármaco.
o Se utilizan para transportar el fármaco exclusivamente al órgano diana.
- Transporte por ionóforos: los ionóforos son compuestos químicos que se
unen a iones de manera reversible y los transportan.
Planeta Formación y Universidades
4unie
Universidad
Absorción de Fármacos
Concepto de Absorción y Biodisponibilidad
La absorción implica el paso del farmaco desde el lugar de administración hasta la
circulación sistémica, para lo cual debe atravesar las membranas celulares.
La absorción de un fármaco se define utilizando el concepto de biodisponibilidad, que
indica la cantidad de farmaco inalterado que alcanza la circulación sistémica, así como
la velocidad a la que lo hace. Este parámetro depende de la vía de administración y de
los denominados efectos de primer paso, o metabolismo presistémico. Los factores
implicados en el proceso de absorción son:
- Características de la forma farmaceutica.
- Características físico-químicas del fármaco: el grado de ionización, el peso
molecular y la liposolubilidad. - Características del lugar de absorción.
Vías de Administración Enteral
- Vías de administración enteral:
Vía Oral
- Vía oral:
- Es la vía más fisiológica, la más usada y de elección en tratamientos
crónicos. - La absorción de la mayoría de los fármacos se produce en el intestino
delgado. - Factores que modifican la absorción:
o Valor del pH.
o Vaciamiento gástrico.
o Motilidad intestinal.
o Flujo sanguíneo.
o Presencia de alimentos.
o Acción enzimática.
o Metabolismo presistémico: es la pérdida de fármaco antes de que
alcance la circulación sistémica.
Metabolismo intestinal: la presencia en el epitelio del intestino
delgado de la enzima CYP3A4 da lugar a reacciones metabólicas casi
idénticas a las que tienen lugar en el hígado.
Planeta Formación y Universidades
5unie
Universidad
- Efecto de primer paso hepático: una vez que el fármaco atraviesa la
mucosa intestinal, una parte de él accede directamente al hígado a
través de la vena porta, transformándose en un metabolito inactivo.
Vía Sublingual
- Vía sublingual:
- La utilización de esta vía permite evitar el efecto de primer paso hepático,
el ataque de las enzimas digestivas, el pH gástrico, el metabolismo por la
flora intestinal o las interacciones con los alimentos u otros fármacos. - Su principal limitación es su escasa superficie de absorción.
Vía Rectal
- Vía rectal
- La absorción tiene lugar en la mucosa rectal por difusión pasiva.
- Es una zona muy vascularizada, con lo que se evita un posible efecto de
primer paso hepático. - Se suele utilizar para conseguir efectos locales.
- Se emplea como alternativa a la vía oral ante la presencia de vómitos,
intolerancia gastrointestinal o pacientes poco colaboradores.
Vías de Administración Tópica y a Través de Mucosas
- Vías de administración tópica y a través de mucosas
Vía Dérmica
- Vía dérmica:
- Se quiere conseguir:
o Un efecto local o tópico.
o Un efecto sistémico, lento y sostenido por absorción percutánea.
- Solo los fármacos muy liposolubles y de bajo peso molecular serán
administrados por esta vía. - Para favorecer este paso, las formulaciones farmacéuticas utilizadas
contienen excipientes muy grasos.
Vías de Administración Parenterales o Directas
- Vías de administración parenterales o directas
Vía Intravenosa
- Vía intravenosa
- El fármaco se administra directamente en el torrente circulatorio.
- No hay absorción y la biodisponibilidad es del 100%
- Solo se pueden administrar soluciones acuosas estériles, libres de
pirógenos e isotónicas.
Planeta Formación y Universidades
6unie
Universidad
- Riesgos: extravasación o infiltración.
Otras Vías Parenterales
- Otras vías parenterales:
VÍA DE ADMINISTRACIÓN
CARACTERISTICAS
Intradérmica
No hay absorción sistémica
La absorción es lenta
Subcutánea
Absorción rápida si se utilizan soluciones
acuosas.
Se debe evitar en pacientes con:
- Trastornos circulatorios
- Patologías de la piel
Puede provocar necrosis y dolor
Intramuscular
Rápidamente
se
consiguen
concentraciones en sangre si se utilizan
soluciones
acuosas
Menos
dolorosa
Se debe evitar en pacientes:
- En tratamiento con anticoagulante
- Con signos de hipoperfusión tisular
Distribución de Fármacos
Factores que Afectan la Distribución
Tras acceder a la circulación sistémica, el fármaco se «reparte» por todo el organismo
transportado en la sangre. Una vez en sangre, los fármacos pueden unirse a proteínas
plasmáticas, en un determinado porcentaje, o a células sanguíneas, principalmente
eritrocitos. El fármaco libre, que no se ha unido, puede atravesar las diferentes
membranas celulares, principalmente por difusión pasiva.
- Factores que afectan a la distribución:
Características Físico-Químicas del Fármaco
- Características físico-químicas del fármaco:
- A mayor liposolubilidad y menor tamaño molecular, mayor será su
distribución.
Planeta Formación y Universidades
7unie
Universidad
Unión del Fármaco a Proteínas Plasmáticas
- Unión del fármaco a las proteínas plasmáticas: un fármaco puede circular en el
torrente sanguíneo disuelto en el plasma (fracción libre) o unido de forma
reversible a las proteínas plasmáticas.
- El porcentaje de unión es variable (0-99%), y se considera una constante
para cada fármaco, independiente de su concentración plasmática y de la
vía de administración. - La fracción que se encuentra unida (complejo fármaco-proteína) es
farmacológicamente inactiva, actuando como un reservorio. - La fracción libre se distribuye a los tejidos, puede alcanzar la célula diana
(ejerce la acción farmacológica) y se elimina. - Ambas formas se encuentran en equilibrio.
- La unión a proteínas plasmáticas determina el volumen aparente de
distribución (Vd). - El grado de unión a proteínas plasmáticas determina el período de latencia,
su intensidad y su duración.
Proteína plasmática
Fármaco libre
Fármaco unido
Órgano diana
A
Fármaco con alta afinidad
Fármaco con baja afinidad
Órgano diana
B
Flujo Sanguíneo y Afinidad Tisular
- Flujo sanguíneo: A causa del tropismo tisular, se produce una redistribución
del fármaco, es decir, el fármaco llega primero a los tejidos más irrigados,
posteriormente, se distribuye a los tejidos menos vascularizados.
Planeta Formación y Universidades
8unie
Universidad
Afinidad del farmaco por el tejido: Algunos tejidos tienen la capacidad de
acumular y almacenar los fármacos tras su absorción, actuando como
reservorios.
Barreras Especiales
- Barreras especiales: La existencia de barreras especiales protege a los órganos
de acceso restringido del paso de sustancias potencialmente tóxicas.
o Barrera hematoencefálica (BHE): es la menos permeable. La BHE está
formada por capilares continuos, con células endoteliales íntimamente
unidas, constituyendo una estructura protectora muy poco permeable
al paso de los fármacos.
· Solo atraviesan por difusión pasiva moléculas pequeñas y muy
lipófilas
o Barrera placentaria: constituida por el endotelio capilar y el trofoblasto
de las vellosidades placentarias, permite el acceso al feto de la mayoría
de los fármacos administrados a la madre durante la gestación o en el
momento del parto.
o Barrera testicular.
Volumen Aparente de Distribución
- Volumen aparente de distribución: volumen de líquido en el que está disuelto el
fármaco cuando se ha alcanzado el equilibrio de distribución.
o Relaciona la dosis de farmaco administrada con la concentración
plasmática máxima, alcanzada tras administración intravenosa.
o Se expresa en litros.
o La magnitud será dependiente de la liposolubilidad del fármaco, de su
afinidad por las proteínas plasmáticas o tisulares, de la perfusión tisular
y de la existencia de barreras especiales.
· Fármacos poco liposolubles que solo alcanzan concentraciones en
sangre y/o con alta unión a proteínas presentarán Vd bajos.
Fármacos muy liposolubles y con bajo grado de unión a proteínas
presentarán Vd elevados.
Planeta Formación y Universidades
9
¿Non has encontrado lo que buscabas?
Explora otros temas en la Algor library o crea directamente tus materiales con la IA.
