Malformaciones Congénitas Craneoencefálicas y Tumores del SNC
Documento de Universidad sobre Malformaciones Congénitas Craneoencefálicas. El Pdf explora disrafismos, anomalías de migración neuronal y malformaciones de la charnela occipital, además de tumores del SNC. Este material de Biología es útil para el estudio autónomo, proporcionando explicaciones detalladas y clasificaciones.
Ver más18 páginas


Visualiza gratis el PDF completo
Regístrate para acceder al documento completo y transformarlo con la IA.
Vista previa
Malformaciones Congénitas Craneoencefálicas
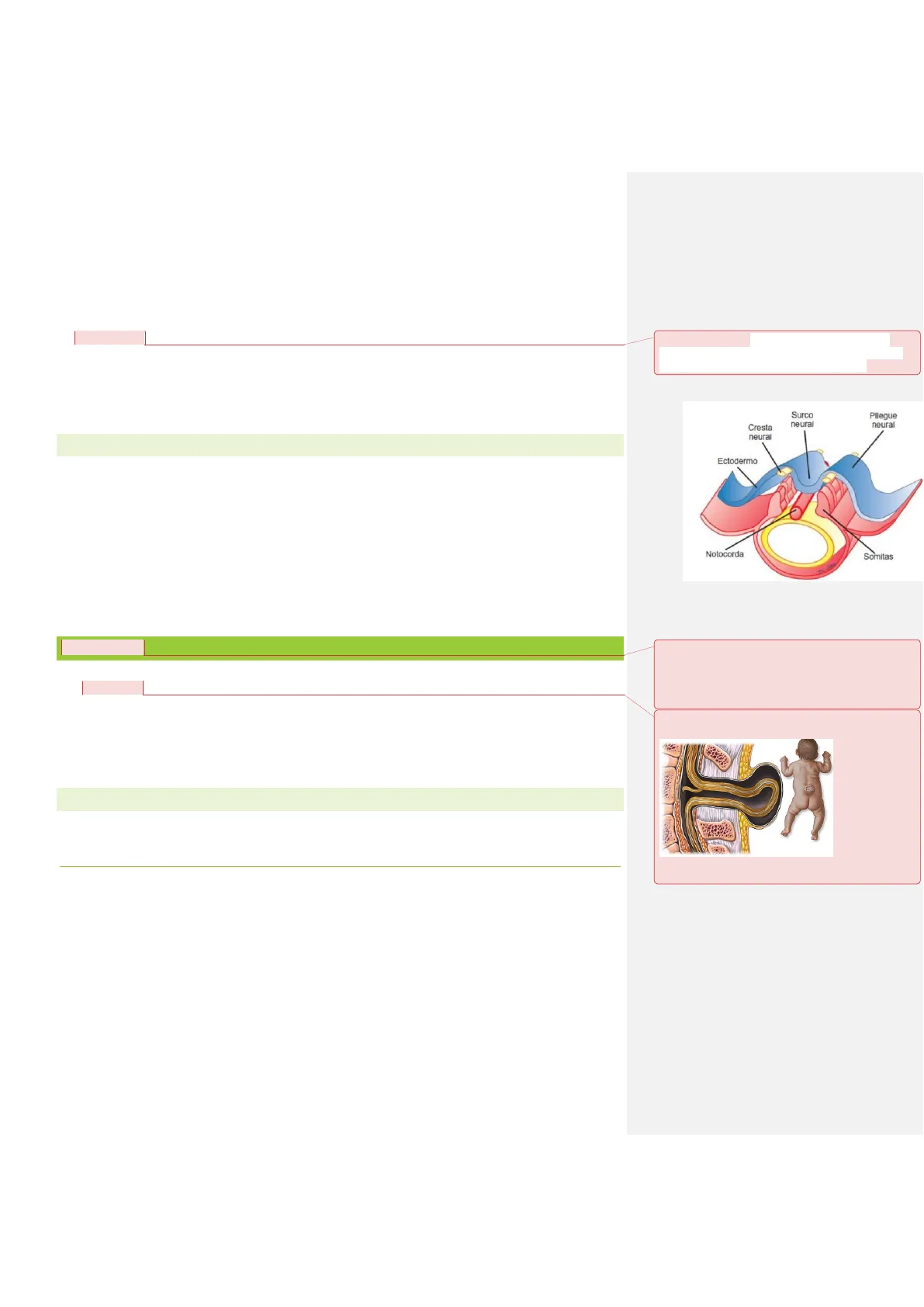
La organogenesis del SNC puede finalizar incluso después del nacimiento, por lo que estas malformaciones pueden darse en distintas etapas del desarrollo, y dependiendo del momento, serán más o menos importantes.
- 1º trimestre del embarazo > las lesiones dificultan la formación del tubo neural.
- 2º trimestre >perturban la proliferación y migración neuronal (lisencefalia, paquigiria, microgiria, heterotopias).
- 3º trimestre > afectan a la organización neuronal y la mielinización (esquisencefalia, poroencefalia, hidranencefalia,)
Clasificación de Malformaciones
- Alteraciones del tubo neural
- Disrafismos > espina bifida
- Hidrocefalia
- Anomalías de la migración neuronal >lisencefalia
- Alteraciones línea media cerebral > agenesia o disgenesia del cuerpo calloso
- Anomalias de la charnela occipital
- Craneosinostosis
- Disostosis craneofacial (Sd. Crouzon)
- Síndromes neurocutáneos >facomatosis
- Vasculares > aneurismas y malformaciones arteriovenosas.
Disrafismos
Los defectos del tubo neural son defectos congénitos del cerebro y la médula espinal.
- Espina bifida: la columna vertebral del feto no se cierra completamente durante el 1º mes de embarazo. Suele haber daño neurológico.
- Anencefalia: gran parte del cerebro no se desarrolla. Los bebes con anencefalia nacen muertos o mueren poco tiempo después
- Encefalocele: apertura en el cráneo a través de la cual hay una protuberancia del tejido cerebral. Pocos sobrevivientes.
Espina Bífida
La columna vertebral del feto no se cierra completamente durante el 1º mes de embarazo, para cubrir la ME, n. raquídeos y las meninges . Suele haber daño neurológico. CAUSA: desconocida.
Tipos de Espina Bífida
- Raquisquisis: apertura completa del canal raquídeo y falta también la parte posterior de médula, meninges y vértebras. Puede asociarse a anencefalia. Es la forma más grave y suelen morir en pocas horas o días.
- Mielomeningocele
- Meningocele: en el quiste solo hay meninges y LCR. No suele cursar con hidrocefalia.
- Espina bífida oculta: en las que el defecto del tubo neural está recubierto por piel. Es asintomática y el diagnóstico es un hallazgo casual al hacer una Radiografía
Mielomeningocele
Es la forma más frecuente de presentación y puede afectar a cualquier parte de la columna, aunque es más frecuente a nivel lumbosacro. Aparece un quiste que contiene meninges, médula y/o raíces nerviosas.
- ETIOLOGÍA: Desconocida. Parece deberse a un déficit de folatos en la madre momentos antes de producirse el embarazo, o si la madre toma ácido valproico, carbamacepina y medicamentos hormonales.
- CLÍNICA: dependerá del nivel. El recién nacido tendrá paresia o parálisis flácida, arreflexia y pérdida de la sensibilidad termoalgésica desde el nivel lesional hacia abajo. Alteraciones esfinterianas y atrofias musculares. Î riesgo de meningitis. Hidrocefalia (90%) y malformación de Arnold-Chiari II (70%)
- DIAGNÓSTICO: Por ecografía en época prenatal, en sangre y líq. amniótico; 1 niveles de alfa-fetoproteína; en TC y RM
- TRATAMIENTO: quirúrgico para cerrar el defecto y de la hidrocefalia para hacer derivación ventriculoperitoneal
Anomalías de la Migración Neuronal
Lisencefalia
LISENCEFALIA: las neuronas al migrar, no completan su trayecto cortical. La superficie cerebral tendrá menos surcos, con engrosamiento de la corteza cerebral. Se suele asociar a disgenesia del cuerpo calloso. Existen distintos grados y la clínica es variable; casi siempre hay retraso mental
Alteraciones de la Línea Media
Agenesia del Cuerpo Calloso
Agenesia del cuerpo calloso: aislada o asociada a otros síndromes como en la trisomía 13 y la 18. CLÍNICA: variable, retraso mental, epilepsia y parálisis cerebral.
Malformaciones de la Charnela Occipital
Síndrome de Klippel Fiel
Consiste en la fusión de varias vértebras cervicales, pero no produce síntomas neurológicos. Los pacientes con estas malformaciones suelen tener el cuello corto, con limitación de movimientos.
Malformación de Dandy-Walker
Consiste en la agenesia total o parcial del vermis cerebeloso con dilatación del IV ventrículo y alargamiento de las estructuras de la fosa posterior. Atresia de foramenes con obstrucción e hidrocefalia. Se puede asociar a otras malformaciones en un 68% de los casos
- CLÍNICA: retraso del desarrollo motor, aumento del tamaño del cráneo del niño, irritabilidad, vómitos, convulsiones, falta de coordinación.
- DIAGNÓSTICO: por la clínica y RM
- TRATAMIENTO: quirúrgico y derivación ventrículo peritoneal para corregir la hidrocefalia
Malformación de Arnold Chiari
Localizada en la unión entre el encéfalo y la ME, localizada en la fosa posterior/base del cerebro. Pertenece al grupo de las malf. de charnel.
- DIAGNÓSTICO DEL ARNOLDO CHARI: RM que demuestra el desplazamiento del cerebelo y puede detectar una siringomielia.
- TRATAMIENTO: descompresión quirúrgica su occipital. Con frecuencia hay que hacer una derivación ventriculoperitoneal para tratar la hidrocefalia.
Arnold Chiari I
Cuando hay un leve descenso de las amígdalas cerebelosas por debajo del foramen magnum, que puede acompañar de descenso del bulbo. Se puede asociar a siringomielia. SÍNTOMAS: en la adolescencia o más tarde (cefalea recurrente y espasticidad)
Arnold Chiari Tipo II
descenso de las amígdalas, el vermis, IV ventrículo y tronco cerebral. Se asocia casi siempre al mielo meningocele e hidrocefalia progresiva. SÍNTOMAS: primeros meses de vida (llanto débil, dificultad para tragar, respirar, pérdida de fuerza, alteraciones de la sensibilidad)
Craneosinostosis
Es el cierre prematuro de una o varias suturas craneales, el craneo deja de crecer zonas sinostosadas y crece por donde el cráneo todavía no está sinostosado. Se manifiesta por deformidad craneal. Puede ser un hecho aislado o formar parte de un Sd. con otras malformaciones asociadas. Si la sinostosis es multiple -> hipertensión intracraneal, microcefalia y déficit intelectual; si no es múltiple, el crecimiento cerebral es normal y no hay déficit intelectual. El trastorno suele ser esporádico (por azar) aunque en algunas familias se hereda (ad o ar).
- CLÍNICA DE LAS CRANEOSINOSTOSIS: Los bebes con craneosinostosis presentan cambios en la forma de la cabeza y la cara, que suelen ser evidentes. Pueden tener fontanela abultada o cerrada, somnolencia, aumento de irritabilidad, llanto fuerte y agudo, vómitos explosivos, convulsiones, etc.
- DIAGNÓSTICO: por examen físico y por: Rx cabeza, Tc.
- TRATAMIENTO: según el tipo y los problemas que acarree. Casi siempre será quirúrgico.
Escafocefalia o Dolicocefalia
ESCAFOCEFALIA O DOLICOCEFALIA: Cierre precoz de la sutura sagital. El resultado es una cabeza alargada en sentido anteroposterior. No produce hipertensión; solo un problema estético. Más frecuente.
Trigonocefalia o Cráneo en Cuña
TRIGONOCEFALIA O CRÁNEO EN CUÑA: Cierre en la sua from. fr / se aprecia hipotelorismo (ojos anormalmente juntos). Únicamente estético.
Plagiocefalia Posterior
PLAGIOCEFALIA POSTERIOR: C. sutura lambdoidea. Muy poco frecuente. Se produce un aplanamiento posterior de uno de los huesos occipitales. Es importante diferenciarla con la plagiocefalia posicional o postural del lactante.
Plagiocefalia Posicional
PLAGIOCEFALIA POSICIONAL: Aplanamiento del cráneo sobre la sutura lambdoidea o la región posterior del cráneo durante la vida intrauterina (posiciones fetales prolongadas, embarazos múltiples, anomalías uterinas, macrocefalia, partos con fórceps ... ) o posterior (por posición mantenida del neonato, tortícolis, posturas determinadas). Clínica:aplanamiento y alopecia en la región occipital, abombamiento contralateral y pabellón auricular adelantado. A veces se asocia a tortícolis, macrocefalia o hidrocefalia. Ø del cráneo: leve (0-10 mm), moderada (11-12) o severa (>20)
Síndromes con Craneosinostosis
Disostosis Craneofacial o Síndrome de Crouzon
DISOSTOSIS CRANEOFACIAL O SÍNDROME DE CROUZON: los huesos del cráneo y del rostro se unen demasiado temprano. Poco frecuente
- SINTOMAS: proptosis ocular por órbitas poco profunda, con o sin estrabismo, hipertelorismo, abultamiento frontal, hipoplasia de los maxilares con o sin nariz en curva. Puede asociarse con hidrocefalia, anomalías dentales y pérdida de audición.
- DIAGNÓSTICO: Rx, RM y estudios genéticos.
- TRATAMIENTO: sintomático
Síndromes Neurocutáneos o Hamartoblastosis
SINDROMES NEUROCUTÁNEOS O HAMARTOBLASTOSIS -> FACOMATOSIS: Grupo de enfermedades hereditarias de origen desconocido del plasma germinativo, con tendencia a la aparición de malformaciones, manchas muco-cutáneas y tumores en numerosos órganos, entre los que destacan la piel y el SNC. Son displasias o anomalías del desarrollo embrionario.
Neurofibromatosis o Enfermedad de Von Recklinghausen
Enfermedad hereditaria.
Tipo 1 Neurofibromatosis
1) TIPO 1 es la más frecuente -> se da por afectación del cromosoma 17
- CLÍNICA del tipo 1 (Neurofibromatosis tipo 1 - NF1):
- Signos mayores: presencia de manchas café con leche (aparecen en el primer año de vida, y deben ser al menos 6) y neurofibromas dérmicos.
- Signos menores: corta estatura y macrocefalia
- Otros sint .: problemas de aprendizaje, TDAH, escoliosis, glioma óptico, neurofibromas en ME, HTA, epilepsia
- COMPLICACIONES del tipo 1:
- Pueden aparecer nódulos en el iris
- Neurofibromas plexiformes: irregulares, voluminosos y se mezclan con el tejido adyacente. A veces hay que extirparlos.
- Cáncer: en un 5% de los casos algún neurofibroma puede degenerar. Tumores embrionarios y neurosarcomas
- DIAGNÓSTICO del tipo 1: clínica y TC
- TRATAMIENTO del tipo 1: sintomático y cirugía de algunos tumores
Tipo 2 Neurofibromatosis
2) TIPO 2: mucho menos frecuente. Aparecen neurinomas bilaterales en el nervio acústico, VIII (hipoacusia). Los síntomas aparecen en la adolescencia tardía
Esclerosis Tuberosa o Enfermedad de Bourneville
Enfermedad hereditaria que aparece los primeros años de la vida de los tumores congénitos y malformaciones en el SN, piel y en en otros órganos.
- INCIDENCIA: rara. Autosómica dominante, a veces esporádica. Afecta a ambos sexos y todas las razas.
- ETIOPATOGENIA: Las lesiones cerebrales mas características son los nódulos corticales o subcorticales de tamaño variable (5 mm-3 cm Ø; tamaño de una cabeza de alfiler/lenteja, color rojizo y sobre todo paranasales) en células gliales y neuronas. Algunos pacientes presentan gliomas retinianos, rabdiomonas en el corazón, , pleura, ovarios, etc., meningiomas ...
- CLÍNICA: suele comenzar con el desarrollo psicomotor y convulsiones durante los dos primeros años de edad. Pueden aparecer lesiones cutáneas faciales en el segundo año de vida.
- TRIADAS CLÁSICA: adenomas sebáceos en cara, epilepsia y retraso mental.
- DIAGNÓSTICO: por la clínica, TC y RM
- TRATAMIENTO: no tratamiento específico, antiepilépticos, resección de los tumores ...
- EVOLUCIÓN: según órganos afectados. Pueden fallecer en primeros años o vivir varias décadas.
¿Non has encontrado lo que buscabas?
Explora otros temas en la Algor library o crea directamente tus materiales con la IA.
