Miocardiopatías: concepto, fenotipos y diagnóstico
Documento de Universidad sobre Miocardiopatías: concepto, fenotipos y diagnóstico. El Pdf detalla los cuatro fenotipos principales (dilatada, hipertrófica, restrictiva y arritmogénica) y sus manifestaciones clínicas. Este documento de Biología es útil para estudiantes universitarios.
Ver más18 páginas

Visualiza gratis el PDF completo
Regístrate para acceder al documento completo y transformarlo con la IA.
Vista previa
TEMA 5: MIOCARDIOPATÍAS
CONCEPTO
Las miocardiopatías son trastornos miocárdicos en los que el músculo cardíaco es estructural y funcionalmente anormal en ausencia de enfermedad arterial coronaria, hipertensión, valvulopatía o cardiopatía congénita suficientes para causar la anomalía miocardica observada.
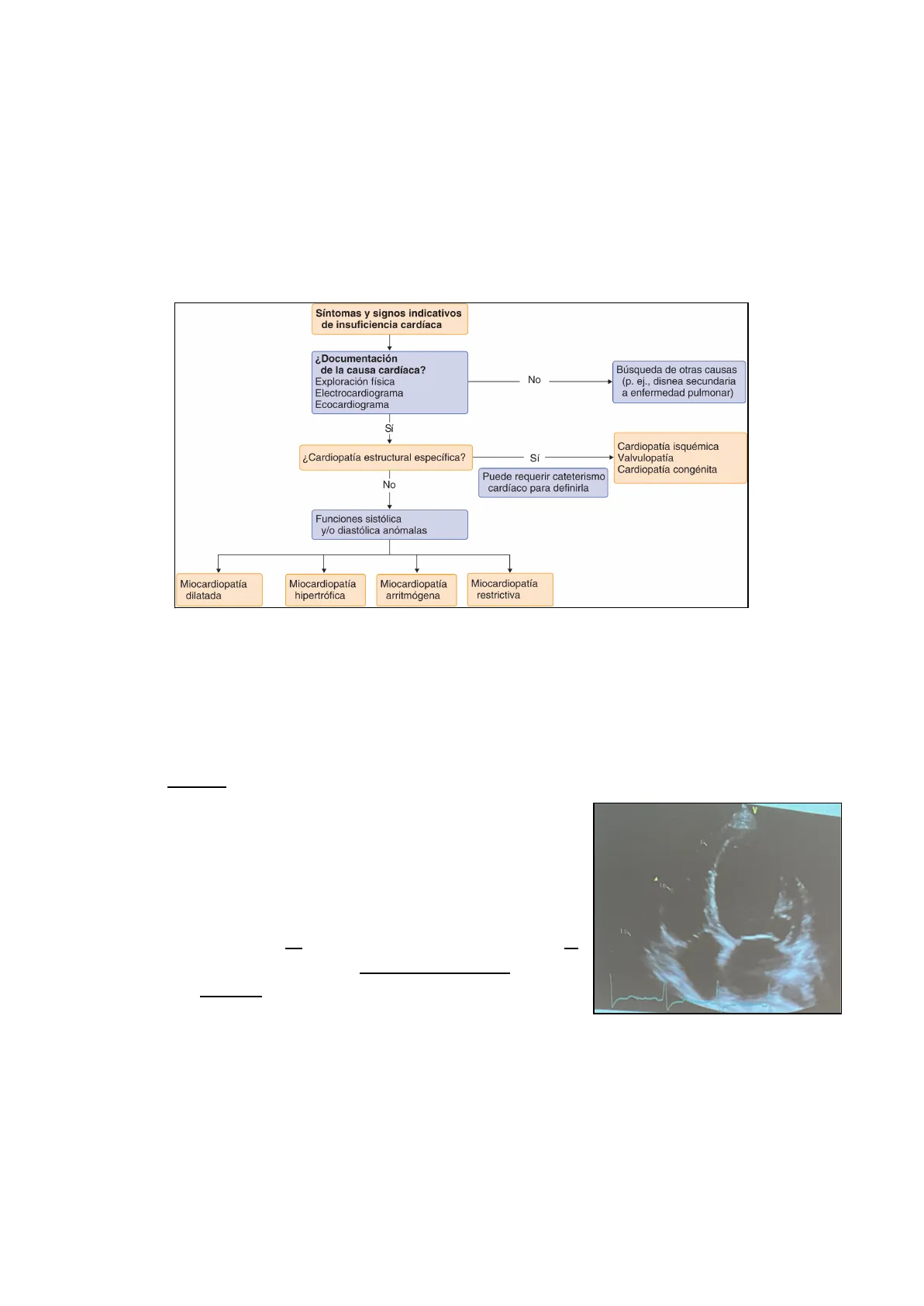
Síntomas y signos indicativos de insuficiencia cardíaca ¿Documentación de la causa cardíaca? Exploración física Electrocardiograma Ecocardiograma No Búsqueda de otras causas (p. ej., disnea secundaria a enfermedad pulmonar) Si ¿Cardiopatía estructural específica? Sí Cardiopatía isquémica Valvulopatía Cardiopatía congénita No Puede requerir cateterismo cardíaco para definirla Funciones sistólica y/o diastólica anómalas Miocardiopatía dilatada Miocardiopatía hipertrófica Miocardiopatía arritmógena Miocardiopatía restrictiva
Aproximación inicial a las miocardiopatías. Clasificación
FENOTIPOS
Nos aproximamos a la miocardiopatías según su fenotipo, diferenciado cuatro tipos principales: dilatada, hipertrófica, restrictiva y arritmogénica de ventrículo derecho (VD).
Miocardiopatía Dilatada
Se parece mucho a la insuficiencia cardíaca con fracción de eyección reducida en ausencia de una causa evidente que la justifique (no hipertensión, infarto ... ). Sus causas principales son la miocarditis (inflamación del corazón), enfermedades metabólicas/endocrinas y causas genéticas.
La fracción de eyección (FE) suele estar reducida, la cavidad del VI en diástole suele estar dilatada, el grosor de las paredes suele ser normal y las aurículas están dilatadas (si hay disfunción sistólica, suele haber disfunción diastólica _asociada). Si encontramos alguna valvulopatía suele ser de tipo funcional mitral o tricuspídea.
La presentación clínica se parece a la de la insuficiencia cardiaca con disnea, cansancio, ortopnea, disnea paroxística nocturna; y pueden producirse arritmias, sobre todo ventriculares, y en algunas causas específicas, trastornos de la conducción, como en la enfermedad del Chagas, miocarditis de células gigantes o laminopatías.
Miocardiopatía Hipertrófica
97B. Hipertrófica Predomina el engrosamiento o hipertrofia de la pared del VI, el cuál tendrá un tamaño normal o reducido. La FE o capacidad de contracción es normal o exagerada (hipercontractilidad), hay disfunción diastólica con las aurículas dilatadas.
Generalmente es de causa genética o puede haber enfermedades infiltrativas o de depósito en el miocardio.
La presentación clínica incluye cansancio, disnea, alteración en el ECG, palpitaciones, y en fases avanzadas, presenta ortopnea y disnea paroxística nocturna. De valvulopatías pueden presentar insuficiencia mitral (SAM), y en cuanto a las arritmias, la fibrilación auricular (FA) suele ser habitual en fases avanzadas. También pueden presentar taquicardia ventricular, bloqueo de la conducción en PRKAG2, mitocondrial, enfermedad de Fabry.
Miocardiopatía Restrictiva
c. Restrictiva Tiene un VI de tamaño normal con el grosor de las paredes normal o ligeramente aumentado con la FE normal. Predomina la disfunción diastólica acompañada de una intensa dilatación auricular.
La causa más frecuente es la amiloidosis (IMPORTANTE), que es una enfermedad infiltrativa por depósito de una proteína anómala), aunque también puede haber causas genéticas o endomiocardicas.
Presenta síntomas propios de insuficiencia cardíaca con predominio de congestión sistémica (disnea y más adelante ortopnea, DPN, insuficiencia cardíaca derecha). En cuanto a las valvulopatías Insuficiencia mitral y tricuspídea, aunque rara vez son graves; y puede haber arritmias con la FA, bloqueo de la conducción en sarcoidosis, amiloidosis o desminopatía.
Miocardiopatía Arritmogénica de VD
D. Arritmogénica de VD Se caracteriza por la sustitución de la pared libre del VD por tejido adiposo y fibrosis, siendo su causa genética (autosómica dominante).
Los pacientes presentan una FE normal o levemente reducida con un tamaño ventricular normal o levemente aumentado en fases avanzadas. El VI tiene un grosor normal y las aurículas son normales hasta el estadio final.
La presentación inicial va a ser en forma de arritmia y, sólo en fases muy avanzadas, se presenta clínica de insuficiencia cardíaca de predominio derecho; con arritmias como ectopia biventricular y taquicardia.
HIPERTRÓFICA DILATADA RESTRICTIVA ARRITMOG. VD
Causas Genética Enf. infiltrativa Miocarditis Metabólicas/endocrinas Genética Enf. infiltrativa Endomiocardicas Genética Genética
FE del VI î o normal ↓ Į o normal normal hasta fase avanzada V
10. 98Tamaño diastolico VI ↓ ↑ normal normal hasta fase avanzada
Grosor VI ↑ normal normal o 1 normal
Tamaño aurículas ↑ ↑ muy î normal hasta fase avanzada
Valvulopatías Insuf. mitral (SAM) Mitral (funcional) Insuf. tricuspidea Rara vez graves
Síntomas Disnea; dolor torácico, síncope, palpitaciones, cansancio Más adelante: ortopnea, DPN Disnea, cansancio Más adelante: ortopnea, DPN Disnea Más adelante: ortopnea, DPN, insuficiencia cardíaca derecha Palpitaciones, síncope Estadio final: insuficiencia cardíaca derecha y/o izquierda
Arritmia Fibrilación auricular, taquicardia ventricular; bloqueo de la conducción en PRKAG2, mitocondrial; enfermedad de Fabry Taquiarritmias ventriculares; bloqueo cardíaco en la enfermedad de Chagas, miocarditis de células gigantes, laminopatías Fibrilación auricular; bloqueo de la conducción en sarcoidosis, amiloidosis, desminopatía Ectopia biventricular y taquicardia
MCH MCD MCNDVI MAVD MCR
(NO IMPORTANTE) Recomendaciones recientes de las guías europeas tienen en cuenta un quinto fenotipo, dada la existencia de disfunción sistólica sin dilatación del VI, así como alteraciones segmentarias sin causa isquémica del VI. Estos casos se engloban con el nombre de miocardiopatía no dilatada del ventrículo izquierdo. Sin embargo, en nuestro enfoque, estos casos se incluirán dentro de la clasificación de la miocardiopatía dilatada.
PROCESO DIAGNÓSTICO
Ante la presencia de un paciente con sospecha de miocardiopatia, se llevan a cabo los pasos incluidos en la imagen. Hay que tener en cuenta que la historia familiar es muy importante, así como los antecedentes de exposiciones a infecciones, drogas o tratamiento quimio/radioterápico.
Además, los test genéticos y la resonancia cardíaca (que permite caracterizar muy bien el miocardio) son herramientas muy útiles.
99Miocardiopatías
1er Paso Diagnóstico
Entrevista clínica: historia clínica + historia familiar ≥ 3 generaciones. Potenciales causas: drogas, infecciones Laboratorio: hemograma, función renal, hepática, hormonas tiroideas, metabolismo del hierro, biomarcadores Radiografía de tórax
2° Paso Diagnóstico
Ecocardiograma Estructura y función cardíaca, identificar fenotipo Identificar características sugestivas de etiología específica Excluir origen isquémico, valvular o hipertensivo
3er Paso Diagnóstico
Estudios complementarios para evaluar sospecha diagnóstica
Disfunción ventricular Función conservada con grosor parietal normal Función ventricular conservada con grosor parietal aumentado
MCD (MCH o MCR en estadio terminal) MCR MCH MCR
RMN para caracterización tisular Test genético RMN para caracterización tisular Test genético RMN para caracterización tisular Test genético
Anderson-Fabry: a-galactosidasa Descartar infecciones (serologías) Descartar enefermedad endocrina o metabólica (hormonas tiroideas, hormona de crecimiento, catecolaminas) Amiloidosis: electroforesis, cadenas ligeras, biopsia grasa Sarcoidosis: niveles ACE, PET Sobrecarga de hierro Amiloidosis: electroforesis, cadenas ligeras, biopsia grasa Sarcoidosis: niveles ACE, PET Sobrecarga de hierro
La RM cardíaca permite evaluar la presencia de fibrosis y de cicatrices en el espacio extracelular usando gadolinio. Cuando se inyecta, se extravasa al espacio extracelular y no se lava/elimina, mientras que en áreas de miocardio normal, sí se elimina. (NO IMPORTANTE) Esto permite identificar, en una secuencia tardía, el "realce tardío de gadolinio", que muestra las cicatrices características de la cardiopatía isquémica (patrones de cicatrices subendocárdicas o transmurales). Además, puede revelar otros patrones con distribución diferente que pueden ayudar a identificar las características específicas de la miocardiopatía.
Patrón isquémico B Intramiocárdico Patrón no isquémico Subepicárdico Subendocárdico circunferencial C E G D
Patrones de realce tardío de gadolinio. A. Infarto subendocardico. B. Infarto transmural. C. Realce lineal intramiocárdico septal. D. Realce en puntos de inserción septal del ventrículo derecho. E. Intramiocárdico inferolateral basal: se encuentra en miocarditis, sarcoidosis, enfermedad de Fabry entre otros. F. Subepicárdico. G. Subendocárdico circunferencial, típico de amiloidosis.
MIOCARDIOPATÍA DILATADA
100 A4. MIOCARDIOPATÍA DILATADA Es un trastorno del músculo cardíaco que se define por la dilatación y disfunción ventricular en ausencia de los culpables habituales (coronariopatía, valvulopatia o enfermedad del pericardio).
En adultos, la incidencia estimada es de 1 cada 250. En conjunto, hombres y mujeres se ven afectados por igual, excepto cuando la herencia está ligada al cromosoma X o a genes mitocondriales.
ETIOLOGÍA de la Miocardiopatía Dilatada
Causas genéticas de Miocardiopatía Dilatada
Entre el 30-50% de las personas con miocardiopatía dilatada presentan indicios de enfermedad familiar (miocardiopatías idiopáticas). En los estudios genéticos, se ve que hay mutaciones en determinados genes del sarcómero.
La herencia autosómica dominante (97%) adopta dos fenotipos principales: miocardiopatia dilatada aislada, que se manifiesta con un cuadro clínico de insuficiencia cardíaca, y miocardiopatia dilatada con arritmia asociada, que puede provocar insuficiencia cardíaca o estar asociada a una miopatía esquelética.
La causa más frecuente son mutaciones en la proteína tinina (TTN, IMPORTANTE) del sarcómero; aunque también pueden aparecer mutadas proteínas del citoesqueleto como la lamina A/C o la filamina C, que se asocian a miocardiopatías dilatadas y que tienen especial tendencia a manifestarse con arritmias ventriculares y muerte súbita.
Las formas de herencia ligada al cromosoma X (3-5%) están principalmente asociadas a distrofias musculares (27%). La miocardiopatía aislada, en estos casos, también se debe a mutaciones del gen de la distrofina, y se caracteriza por la elevación de las isoformas de la creatina cinasa muscular (CK-MB), pero no produce signos ni síntomas de distrofia de la musculatura esquelético.
Causas Adquiridas de Miocardiopatía Dilatada
Todo aquello que haga daño al miocardio y le haga perder miocitos puede acabar dando un patrón de dilatación y disfunción sistólica: miocarditis infecciosa (sobre todo vírica o enfermedad de Chagas), quimioterapia, radioterapia, inmunoterapia, alcohol, cocaína, déficits nutricionales, sobrecarga de Fe, periparto o arritmias (taquimiocardiopatía).
Miocarditis
. Miocarditis La miocarditis es un proceso inflamatorio de origen infeccioso, tóxico o inmunomediado que afecta al miocardio.
- Causas infecciosas: la causa más frecuente es viral (virus de Coxsackie, virus de la inmunodeficiencia humana (VIH), ecovirus, síndrome respiratorio agudo grave por coronavirus 2, adenovirus, gripe, sarampión, paperas, parvovirus, poliovirus, rubéola, virus de la varicela-zóster, virus del herpes simple, citomegalovirus, virus de la hepatitis C, virus de la rabia, virus sincitial respiratorio, virus vacuna, virus del dengue, virus de la fiebre amarilla), aunque también puede ser protozoaria (Trypanosoma cruzi, Toxoplasma gondii), bacteriana (Brucella, Corynebacterium diphtheriae, Salmonella, Haemophilus 101
¿Non has encontrado lo que buscabas?
Explora otros temas en la Algor library o crea directamente tus materiales con la IA.
