Fibrosi Cistica: eziologia, patogenesi, diagnosi e terapie
Slide della Prof.ssa Missiroli sulla fibrosi cistica, una malattia genetica. Il Pdf, utile per lo studio universitario di Biologia, descrive la patologia, le manifestazioni cliniche, le metodologie diagnostiche e le opzioni terapeutiche, arricchito da schemi e grafici.
Mostra di più11 pagine


Visualizza gratis il Pdf completo
Registrati per accedere all’intero documento e trasformarlo con l’AI.
Anteprima
Fibrosi Cistica: Panoramica
Patologia I ed Immunologia, Lezione 5, 11/03/24 Prof.ssa Missiroli FIBROSI CISTICA La fibrosi cistica, scientificamente nota come mucoviscidosi, è una patologia genetica che comporta la produzione di muco denso, viscoso e disidratato, con conseguente formazione di cisti fibrotiche. È una malattia relativamente frequente, soprattutto nella popolazione di razza caucasica, con un'incidenza di circa 1 su 2.500 nati vivi.
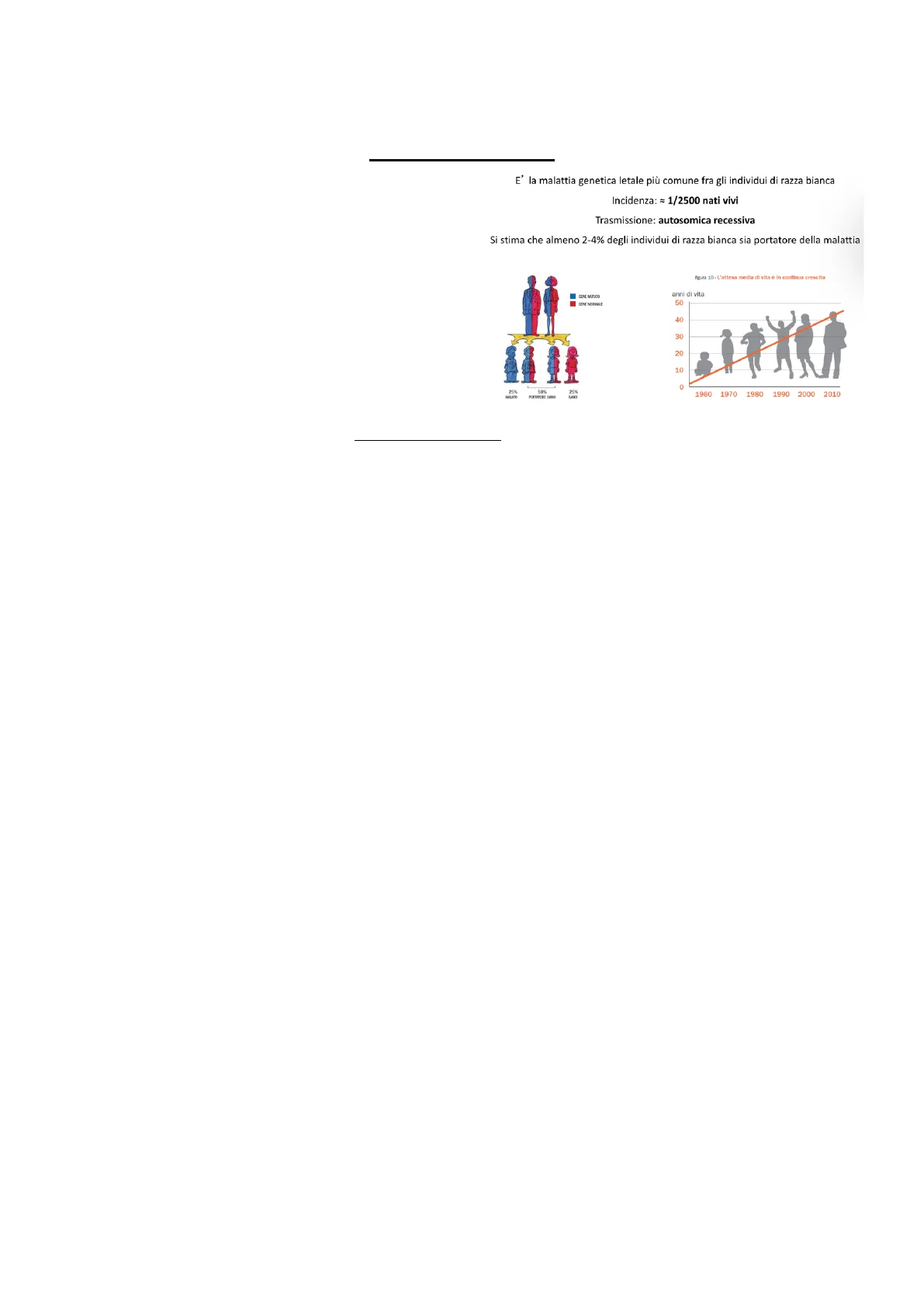
E' la malattia genetica letale più comune fra gli individui di razza bianca Incidenza: = 1/2500 nati vivi Trasmissione: autosomica recessiva Si stima che almeno 2-4% degli individui di razza bianca sia portatore della malattia
GENE MUTADO GENE NORMALE 25% 50% PORTATORE SANO 25K SINO figura 10- L'attesa media di vita è in continua crescita anni di vita 50 40 30 20 10 O 1960 1970 1980 1990 2000 2010
La trasmissione della fibrosi cistica è autosomica recessiva, il che significa che entrambi i genitori devono essere portatori del gene mutato per la malattia. Si stima che circa 1 su 25 individui sia portatore del gene mutato. Se entrambi i genitori sono portatori, esiste una probabilità del 25% che il loro figlio erediti entrambe le copie mutate del gene e sviluppi la malattia.
Le aspettative di vita per i pazienti affetti da fibrosi cistica sono notevolmente migliorate nel corso degli anni. In passato (negli anni 50/60) la malattia era considerata pediatrica poiché le aspettative di vita erano molto brevi. Tuttavia, grazie agli avanzamenti nella ricerca e alla comprensione della causa genetica della malattia, le aspettative di vita sono aumentate significativamente, consentendo a molti pazienti di raggiungere l'età adulta. La fibrosi cistica è attualmente una malattia incurabile, ma le terapie disponibili mirano a migliorare la qualità della vita dei pazienti e a ridurre le complicanze.
La fibrosi cistica è una malattia multisistemica che coinvolge diversi organi. Il sistema respiratorio è l'organo maggiormente interessato, seguito dall'apparato digerente, in particolare il pancreas. I pazienti affetti da fibrosi cistica possono sperimentare una serie di manifestazioni cliniche, tra cui problemi respiratori, insufficienza pancreatica, malnutrizione, crisi polmonari e problemi al fegato.
Cosa comporta questo tipo di patologia? Nella mucoviscidosi, le secrezioni mucose sono viscide, dense e disidratate, il che può portare, a lungo andare, all'ostruzione dei dotti principali, causando l'insorgenza delle manifestazioni cliniche tipiche di questa malattia. Quindi, si tratta di una patologia caratterizzata da un'alterata diffusione del processo secretorio che coinvolge tutte le ghiandole esocrine, comprese le ghiandole sudoripare e le ghiandole mucosecernenti, interessando vari distretti del corpo.
Nonostante molti pazienti affetti da fibrosi cistica oggi riescano a raggiungere un'età adulta, la maggior parte di essi soffre di problemi di salute significativi e presenta una qualità di vita compromessa. Le crisi polmonari rappresentano una delle principali cause di mortalità nei pazienti affetti da fibrosi cistica, poiché il polmone è uno degli organi maggiormente colpiti dalla malattia.
Patogenesi Molecolare della Fibrosi Cistica
1Cosa accade dal punto di vista molecolare nella patologia? PATOGENESI MOLECOLARE CFTR: Cystic Fibrosis Transmembrane conductance Regulator (Regolatore della conduttanza Transmembrana della Fibrosi Cistica) Espressione sulla superficie CFTN Regulator, il regolatore della conduttanza OF gine 250 Kb intron Transcription 27 esoni Trafficking Primary transcript delle proteine transmembrana della fibrosi cistica. Il gene del canale RINA processing Gele CFTR si trova sul braccio lungo del cromosoma 7 e CET MANA = 6500 Translation MRNA nucleotidi Traduzione & maturazione delle proteine Chromosome 7 ATP binding sites Reticolo NH COOH Hydrophobic transmembrane regions endoplasmático codifica per una proteina di circa 1500 amminoacidi. CFTR protein 1480 aa Trascrizione 1 genica Una volta trascritta, la proteina deve essere portata al DNA sito di espressione. Si tratta di un canale che si esprime sulle cellule epiteliali ed è fondamentale per la patologia che la proteina venga correttamente trascritta, tradotta, maturata e trasportata al sito di funzionamento.
Funzione del Canale CFTR
CFTR è un canale anionico che consente il passaggio di cloruri e bicarbonati dalla parte interna delle cellule verso l'esterno, ovvero l'ambiente extracellulare. È espresso in quasi tutte le cellule, comprese quelle del sistema immunitario, ma è presente in maggior quantità nelle cellule epiteliali e nelle ghiandole esocrine, dove agisce per estrudere gli ioni cloruro dallo spazio citoplasmatico verso il lume dell'organo. In altre parole, agisce come un canale selettivo che favorisce il movimento degli ioni cloruro verso l'esterno della cellula.
Tuttavia, nelle ghiandole sudoripare, questo canale opera in modo inverso, richiamando gli ioni Cl all'interno della cellula, contribuendo così alla produzione del sudore. Quindi, lo stesso canale svolge due funzioni diverse a seconda del tipo di cellula in cui è espresso. Le ghiandole sudoripare rappresentano l'unico esempio in cui il canale CFTR opera in questo modo inverso; in tutte le altre cellule in cui è espresso, esclude gli ioni cloruro e bicarbonati dalla cellula.
Struttura del Canale CFTR
Come è formato il canale CFTR Il canale CFTR è composto da 5 domini:
- 2 domini trans-membrana (MSD: membrane-spanning domain), ciascuno dei quali è costituito da 6 a- eliche che formano il core del canale. Questi domini presentano un'alta selettività per gli ioni Cl in quanto sono costituiti da domini carichi positivamente.
- 2 domini che legano nucleotidi (NBD: nucleotide- binding domain), localizzati nello spazio citoplasmatico vicino alla membrana plasmatica. Questi domini sono in grado di legare l'ATP, il che significa che per funzionare il CFTR ha bisogno di ATP.
CFTR: Cystic Fibrosis Transmembrane conductance Regulator (Regolatore della conduttanza Transmembrana della Fibrosi Cistica) MSD-1 MSD-2 Extracellular Plasma membrane Intracellular N NBD-1 ATP NBD-2 ADP + P, ADP + P R domain C 1 PKA + ATP 5 domini: 2 DOMINI TRANSMEMBRANA (MSD: membrane-spanning domain) 2 DOMINI CHE LEGANO NUCLEOTIDI (ATP) (NBD: nucleotide-binding domain) requires that CAMP · 1 DOMINIO REGOLATORIO Figure 3. Activity of CFTR as a chloride channel is known to require cyclic AMP, protein kinase A, and ATP, but the details remain unclear. hypothetical scheme, the protein's 12 transmembrane segments form the actual channel. W ctual channel, which in the resting state (top) is plugged by the R-domain. Activation (bottom) requires bigger protein kinase A, which in tum phospho um phosphorylates the domain. The resulting exposure of NEIF's permits binding and hydrolysis of ATP. Uncertainty persists because CFTR is known to function even when all identified phosphorylation sites have been disabled. Moreover, various portions of CFTR exhibit individual chloride conductances. 2
La patologia è caratterizzata da mutazioni del gene CFTR, che codifica per il canale CFTR, acronimo di Cystic Fibrosis Transmembrane Conductance ATF· Dominio regolatorio, situato nello spazio citoplasmatico. È importante per l'apertura del canale e contiene siti di fosforilazione (con serine e treonine) che vengono riconosciuti dalla PKA (proteina chinasi A) e coinvolgono anche il cAMP. Pertanto, una serie di fosforilazioni e il legame con l'ATP permettono che questo anello si stacchi dai domini trans-membrana, consentendo la fuoriuscita degli ioni Cl.
Due meccanismi permettono l'attivazione di questo canale:
- La presenza di ATP.
- La presenza della PKA che riesce a fosforilare i siti bersaglio presenti sul dominio regolatorio.
Mutazioni del Gene CFTR
Ad oggi, sono state descritte più di 2.000 mutazioni coinvolte nel gene per il CFTR responsabili della fibrosi cistica. Nonostante la scoperta del gene risalga al 1989, rendendola piuttosto recente, un'ampia gamma di mutazioni è stata identificata.
Macroscopicamente, possiamo classificare queste mutazioni in due categorie principali:
- Mutazioni che riducono la quantità delle proteine CFTR. Queste mutazioni comportano la mancata sintesi o la riduzione della sintesi del canale, il quale non è espresso o è poco espresso.
- Mutazioni che riducono la funzione delle proteine CFTR. In questo caso, il canale viene tradotto, la proteina può maturare e localizzarsi sulla membrana plasmatica delle cellule epiteliali, ma non è funzionale a causa delle mutazioni presenti.
Il percorso clinico varia a seconda del tipo di mutazione. Le mutazioni che causano la mancata o la riduzione della sintesi del canale hanno ripercussioni più gravi, mentre quelle che portano alla riduzione della funzione del canale hanno ripercussioni meno gravi, poiché il canale è presente ma funziona in modo meno efficiente.
Le 2.000 mutazioni del gene CFTR sono state suddivise in 6 classi:
- Classe 1: mutazioni che portano alla mancata sintesi del canale, come la delezione genica, mutazioni puntiformi sul promotore, mutazioni non-senso o altre mutazioni.
- Classe 2: mutazioni che comportano un blocco nella maturazione del canale dovuto a un'alterazione degli amminoacidi. In questa classe, la proteina viene tradotta ma non riesce a maturare a causa della degradazione precoce da parte del reticolo endoplasmatico. La mutazione più comune in questa classe (presente nel 70% degli infetti) è la delezione della fenilalanina in posizione 508, che causa l'incapacità della proteina di essere glicosilata e la sua rapida degradazione da parte del proteasoma.
Questo tipo di mutazioni, appartenenti alle classi 1 e 2, sono quelle più gravi e comportano una prognosi peggiore per i pazienti.
- Classe 3: Queste mutazioni hanno un impatto sulla funzionalità del canale, rendendolo non funzionale nonostante la presenza della proteina. Un esempio di questo tipo di mutazione avviene nel sito di legame dell'ATP, dove la mutazione impedisce il legame con l'ATP. Anche se il canale è presente, la sua non funzionalità comporta effetti simili a quelli riscontrati nelle classi 1 e 2.
- Classe 4: Queste mutazioni portano a una riduzione della conduttanza del canale. Pur essendo espresso, il canale può perdere la sua selettività per gli ioni cloruro e bicarbonato a causa di cariche negative o amminoacidi idrofobici. Ciò provoca una diminuzione del trasporto di cloruri verso l'esterno.
- Classi 5 e 6: Queste mutazioni comportano una ridotta sintesi o stabilità del canale CFTR. Dal punto di vista clinico, i pazienti affetti da questo tipo di mutazioni hanno una prognosi meno grave. Nei casi di classe 5, il canale è espresso e funzionale ma in numero ridotto, mentre nelle mutazioni di classe 6, il canale è presente e funzionale ma instabile, portando alla sua rapida degradazione da parte del proteasoma. Pertanto, questi tipi di mutazioni conducono a condizioni di vita migliori rispetto alle altre classi.
CFTR nelle Ghiandole Sudoripare e Meccanismo Patogenetico
CFTR nelle ghiandole sudoripare Come detto precedentemente, il canale CFTR nelle ghiandole sudoripare opera in modo inverso, 1. GHIANDOLE SUDORIPARE riassorbendo gli ioni Cl dal sudore. Nei pazienti affetti NORMALE FIBROSI CISTICA LUME GHIANDOLA Skin surface SUDORIPARA CI Na+ Na+ CETE da fibrosi cistica, dove il canale può essere assente, -Sweat duct presente in quantità ridotte o funzionare in modo Area of detail insufficiente, si verifica un mancato riassorbimento degli ioni Cl. Questo fenomeno provoca un'elevata Sweat gland coil concentrazione di sali nel sudore di tali pazienti. Di conseguenza, vi è un aumento degli elettroliti nel sudore dei pazienti con fibrosi cistica, il che rappresenta un punto diagnostico cruciale. Questo test è uno dei più utilizzati in quanto non invasivo, economico e prevede la misurazione degli elettroliti nel sudore. La concentrazione di sali nel sudore può arrivare fino a 70 millimoli per litro.
MECCANISMO PATOGENETICO: Cosa avviene se CFTR non funziona? EFFETTI DIFFERENTI NEI DIVERSI TESSUTI: Decremento del riassorbimento di NaCI > aumento del contenuto di elettroliti nel sudore TEST DIAGNOSTICO: DOSAGGIO DEGLI ELETTROLITI NEL SUDORE > CI aumenta fino a 70 mEq/L (normale 1 mEq/L)
CFTR in tutte le altre cellule Prendiamo ad esempio l'epitelio delle vie aeree: il canale CFTR è espresso nella porzione apicale della membrana plasmatica e ha il compito di estrudere ioni Cl verso lo spazio extracellulare. Questa funzione non è svolta autonomamente, ma lavora in coordinazione con altri sistemi. Il primo con cui collabora è il sistema simporto, localizzato nella parte opposta della cellula (porzione basale). Il sistema simporto agisce per portare gli ioni Cl a livello della parte basale della membrana plasmatica. La presenza di ioni Cl richiama ioni Na+ e 4
Non hai trovato quello che cercavi?
Esplora altri argomenti nella Algor library o crea direttamente i tuoi materiali con l’AI.
