Patologie Mitocondriali: mutazioni del DNA e sindrome di Leigh
Documento del Professor Alberto Danese su Patologie Mitocondriali. Il Pdf, un testo discorsivo di Biologia a livello universitario, esplora le mutazioni del DNA mitocondriale e nucleare, l'eteroplasmia e la sindrome di Leigh, fornendo un quadro completo per lo studio.
Mostra di più13 pagine


Visualizza gratis il Pdf completo
Registrati per accedere all’intero documento e trasformarlo con l’AI.
Anteprima
Patologie Mitocondriali
Patologia, Lezione 9a, 20/03/2023 Professor Alberto Danese PATOLOGIE MITOCONDRIALI Le malattie mitocondriali sono un gruppo molto eterogeneo di patologie. La variabilità riguarda l'età d'insorgenza, i tessuti coinvolti e la progressione. Sono patologie ereditarie caratterizzate da alterazioni nel funzionamento dei mitocondri principalmente a livello della fosforilazione ossidativa, quindi catena di trasporto degli elettroni e di conseguenza produzione di ATP. Possono avere manifestazioni sia organo specifica che multiorgano. Sintomatologicamente parlando, la caratteristica comune è l'intolleranza agli sforzi, il facile affaticamento e l'accumulo di acido lattico nei tessuti muscolari. Le cause sono da ricercare principalmente in mutazioni del DNA mitocondriale e DNA nucleare.
Ripasso sui mitocondri
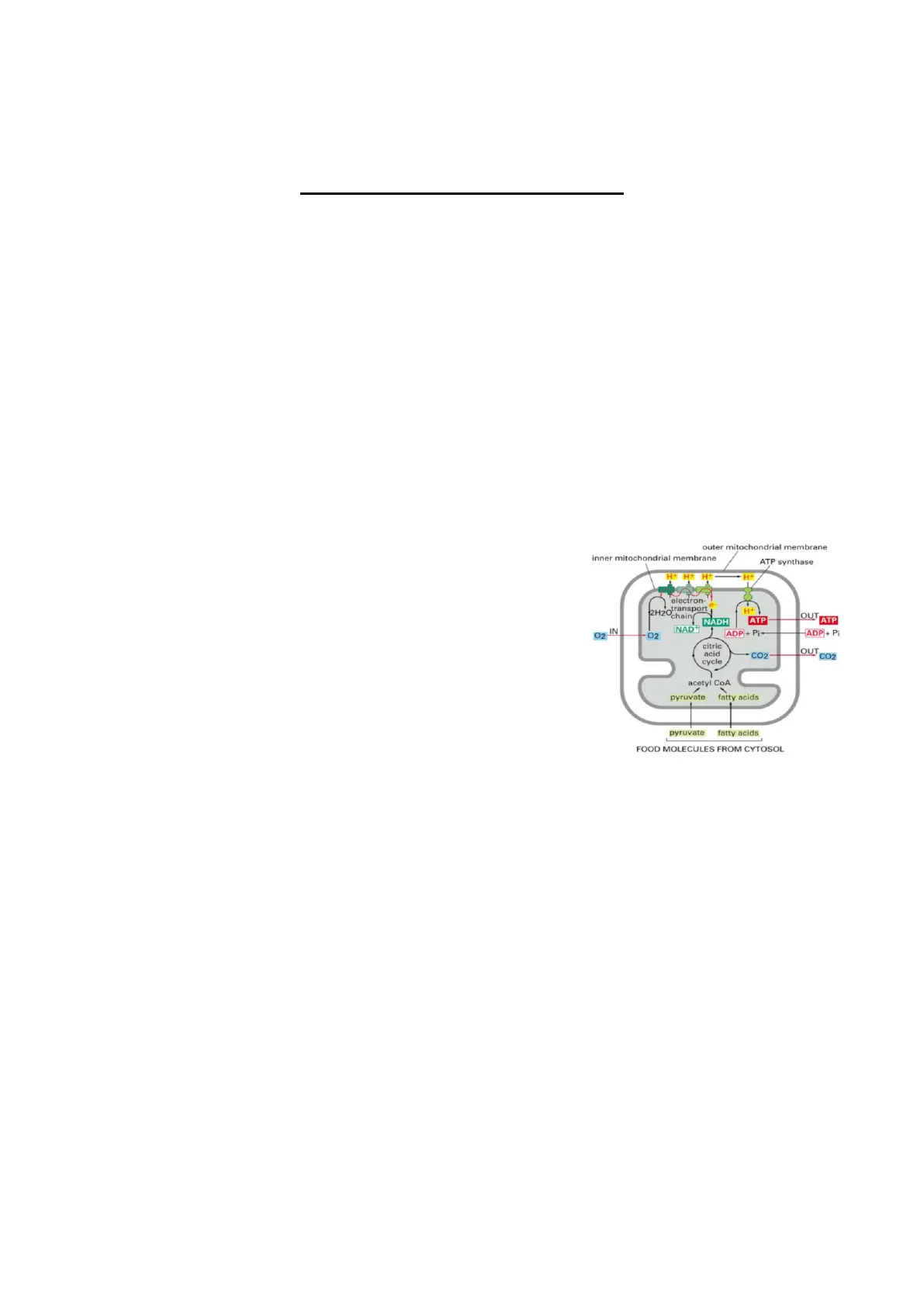
I mitocondri sono organelli cellulari semi autonomi, presenti in tutte le outer mitochondrial membrane inner mitochondrial membrane ATP synthase cellule eucariotiche, sia animali che vegetali, aventi la funzione di produttori di energia principalmente per via ossidativa. Rispetto agli electron- transport H+ 2H2Ochain NADH ATP altri organelli cellulari i mitocondri contengono all'interno della 02 IN -02 NAD+ ADP + Pi- citric acid -CO2 cycle matrice un proprio DNA circolare, definito DNA mitocondriale, il quale probabilmente è il residuo di un organismo procariote acetyl COA 1 pyruvate fatty acids originariamente a sé stante. Questa ipotesi è sostenuta dalla teoria pyruvate fatty acids dell'endosimbiosi, secondo la quale un procariote avrebbe inglobato al FOOD MOLECULES FROM CYTOSOL suo interno il proto-mitocondrio in cambio di reciproci benefici come il metabolismo ossidativo. Difatti possiamo ritrovare le caratteristiche di un procariota: i geni sono privi di introni e vengono trascritti come RNA policistronico ossia che codifica geni a partire da due promotori. Ogni filamento della doppia elica ha un sito di inizio della sintesi di DNA, le sequenze di DNA poste tra i due geni, quindi non ci sono introni ma ci sono sequenze che codificano per RNA transfer. Un'altra cosa molto importante è che il DNA mitocondriale è molto sensibile alle mutazioni soprattutto a carico di stress ossidativo (uno dei prodotti della fosforilazione ossidativa è rappresentato dalle specie reattive dell'ossigeno, i ROS, e il DNA del mitocondrio è molto sensibile a questi radicali). OUT ATP ADP+ Pi OUT CO2
Mutazioni del DNA mitocondriale
Il DNA mitocondriale è il più studiato e a testimonianza di ciò esiste una mappa di tutte le mutazioni patologiche del DNA mitocondriale. La trasmissione si basa sull'ereditarietà materna. Il mtDNA del padre o non viene incorporato nella cellula uovo che darà origine allo zigote o la sua replicazione viene soppressa e il mtDNA del padre viene degradato. Le mutazioni vengono trasmesse dalla madre ai figli. Le mutazioni del mtDNA coesistono con il mtDNA wild-type (quindi sano) creando così le cosiddette cellule eteroplastiche. 1Questa caratteristica è la più comune, ci sono poche situazioni in cui c'è un mtDNA tutto mutato o un mtDNA tutto wild-type. Pertanto la proporzione di mtDNA mutato/wild-type di origine mitocondriale può variare tra i diversi individui e tra tessuti dello stesso individuo.
Eteroplasmia
Definizione di eteroplasmia
ETEROPLASMIA: la presenza di più di un tipo di genoma organellare all'interno di una cellula. Si definisce eteroplasmia la presenza di uno o più tipi di genoma organellare all'interno di una stessa cellula. Questo fenomeno è dovuto ad un Wild type mtDNA WT Mutant mtDNA MUT processo di segregazione casuale dei mitocondri Non disease durante la divisione cellulare per cui le cellule Disease CELL DIVISION figlie avranno un numero di mitocondri pressoché Random segregation uguale e in nessun modo è orchestrato quale mitocondrio debba andare in quale cellula figlia. Quindi la percentuale di eteroplasmia varia a seconda della percentuale di DNA mutato rispetto Normal homoplasmy Heteroplasmy 30% Heteroplasmy 70% Mutant homoplasmy al totale: questo porta una differenza di fenotipo tra cellula e cellula, tra i diversi tessuti e tra i diversi soggetti. Quindi può accadere che in una famiglia ci siano più fratelli e uno sia stato un pochino più fortunato e abbia ereditato mitocondri principalmente wild-type e un fratello più sfortunato ha ereditato più mitocondri mutati quindi eteroplasmia, per ipotesi, al 30% non sviluppa fenotipo malato, al 70 % sono predominanti i mitocondri mutati quindi si sviluppa fenotipo malato. Si parla di omoplasmia sia se ci sono solo mitocondri malati sia se ce ne sono solo di sani. All'interno dei diversi tessuti possiamo avere quindi una differente quantità di mitocondri con il DNA mutato, questa quantità correla direttamente con il grado del difetto biochimico; in alcuni casi queste mutazioni possono essere compensate dal rimanente 5-20% di DNA wild-type. Quindi può essere che una cellula abbia molto poco DNA wild-type, ma sia sufficiente per sopperire ai deficit della mutazione in oggetto sul mtDNA mutato.
Patologie omoplasmiche
Ci sono pochissime patologie mitocondriali che hanno un comportamento omoplasmico; una di queste è la Neuropatia ottica ereditaria di LEBER o meglio detta LOHN. Il DNA mitocondriale non codifica per tutte le proteine responsabili delle funzioni metaboliche del mitocondrio in quanto le proteine della catena respiratoria sono codificate da geni che risiedono nel DNA nucleare, sono quindi poche le proteine codificate dal mtDNA. Difatti la fosforilazione ossidativa richiede più di 60 proteine e la maggior parte di esse sono codificate nel nucleo. Per patologie mitocondriali non si intendono quindi solamente malattie derivanti da mutazioni avvenute a carico del DNA mitocondriale, ma anche malattie che derivano da mutazioni del DNA nucleare che codifica per componenti mitocondriali. La funzione più colpita è appunto la produzione di ATP. 2
Trasmissione
Dato che le malattie mitocondriali possono dipendere sia da alterazioni sul mtDNA che da geni contenuti nel nucleo, la trasmissione può seguire vari percorsi. Quindi solo una piccola parte delle malattie segue le regole classiche dell'eredità: quelle dovute a mutazioni di geni contenuti nel nucleo della cellula e che dunque possono essere sia mutazioni dominanti, sia recessive e sia mutazioni X-linked. Per quanto riguarda la parte mitocondriale, l'eredità è solo materna. XX BR XX 8X XX XX XX XX XX XX 18 17 mtDNA DNA nucleare
Regole di ereditarietà
Regole classiche dell'ereditarietà:
- Mutazione dominante
- Mutazione recessiva
- Mutazione X-linked
Ereditarietà mitocondriale:
- Trasmissione materna
Riassunto sulla trasmissione
Riassunto:
- A livello molecolare le patologie mitocondriali affliggono la funzionalità mitocondriale e di conseguenza la produzione di energia (ATP).
- Sia i geni nucleari, che i geni mitocondriali possono dare origine a difetti del metabolismo energetico, quindi mutazioni sia a carico del DNA nucleare che a carico del DNA mitocondriale possono risultare in patologia.
- Le modalità di trasmissione possono essere due: autosomiche (DNA nucleare) o unicamente materne (mtDNA).
- L'eteroplasmia, dovuta alla segregazione casuale dei mitocondri all'interno della cellula figlia, fa sì che le cellule figlie non posseggano la stessa quantità di mtDNA mutato e che quindi il fenotipo potrà essere diverso da soggetto a soggetto, anche all'interno della stessa famiglia.
- Soltanto le femmine trasmettono il carattere (ereditarietà materna).
- È impossibile prevedere il grado di gravità della malattia sui figli.
Organi e tessuti colpiti
I tessuti maggiormente colpiti sono quelli muscolare e nervoso a causa del loro metabolismo, sebbene i mitocondri si trovino in tutte le cellule; per questo queste malattie vengono definite neuromiopatie mitocondriali o encefalomiopatie mitocondriali. Gli organi maggiormente colpiti sono il sistema nervoso, sistema muscolare, occhi e fegato. Alcune di queste patologie colpiscono un determinato organo, ad esempio la LHON coinvolge sostanzialmente l'occhio. 3
Età di insorgenza
Le manifestazioni cliniche variano durante la crescita dell'individuo:
- Nei neonati (manifestazione grave): grave anemia, disfunzioni del pancreas, del cuore, dei reni e alterazioni dei muscoli.
- Nell'infanzia: rallentamento o arresto della crescita, danni renali, atrofia ottica e sordità, disfunzioni endocrine (ad esempio l'insorgenza precoce del diabete).
- Nell'adulto: il tessuto affetto è principalmente il muscolo scheletrico, di conseguenza abbiamo intolleranza allo sforzo e il facile affaticamento.
Variabilità clinica
La grande variabilità clinica è correlata all'eteroplasmia (quindi alle quantità relative diverse di mitocondri normali e mutati presenti nei diversi tessuti) e questo spiega in parte le differenze nell'espressione delle mutazioni, infatti quando si esegue un test è spesso necessario analizzare più tessuti, poiché, a causa di questo fenomeno, alcuni tessuti possono contenere pochi mitocondri mutanti oppure esserne addirittura privi. Per arrivare alla manifestazione della patologia è necessario superare l'effetto soglia, ovvero il rapporto tra mtDNA mutato e DNA wild-type deve arrivare ad essere a favore della patologia. Quindi una persona può nascere con questo rapporto a favore del fenotipo sano, ma dopo, proprio perché la segregazione casuale dei mitocondri continua per tutta la vita di una persona, vengono selezionati di più i mitocondri mutati rispetto ai mitocondri wild-type. Un ultimo aspetto riguarda la numerosità dei geni colpiti: possono essere colpiti sia geni del mtDNA (37 geni) che del DNA genomico. Complex I Complex Il Complex III Complex IV Complex V 4 H+ 2H+ 2H+ 3H+ Cyta = Cyt b = Com 1 9 Com 2 135.1 Cytochorme C oxidase 2.1.1.2 Succinate dehydrogenase Cytochrom bc1 Complex 7122 72219 7121 ATP synthase inizio spiegazione dell'immagine della catena respiratoria facendone notare la complessità e il fatto che le varie subunitá dei complessi possono essere colpite in vari modi
Classificazione delle malattie
Dal punto di vista medico-scientifico le malattie mitocondriali si possono distinguere in due gruppi nosologici in base alla genetica: 1) Malattie caratterizzate dalla presenza di mutazioni del mtDNA 2) Malattie causate da mutazioni in geni nucleari 4 16993 NADH dehydrogenase 1931
Non hai trovato quello che cercavi?
Esplora altri argomenti nella Algor library o crea direttamente i tuoi materiali con l’AI.
