Neuropatías periféricas y plexopatías de la Universitat Internacional de Catalunya
Diapositivas de la Universitat Internacional de Catalunya sobre neuropatías periféricas y plexopatías. El Pdf explora las plexopatías lumbares por neoplasia, irradiación, diabetes e idiopáticas, con un recuerdo anatómico. Este material de Biología de nivel universitario es útil para el estudio autónomo.
Ver más64 páginas


Visualiza gratis el PDF completo
Regístrate para acceder al documento completo y transformarlo con la IA.
Vista previa
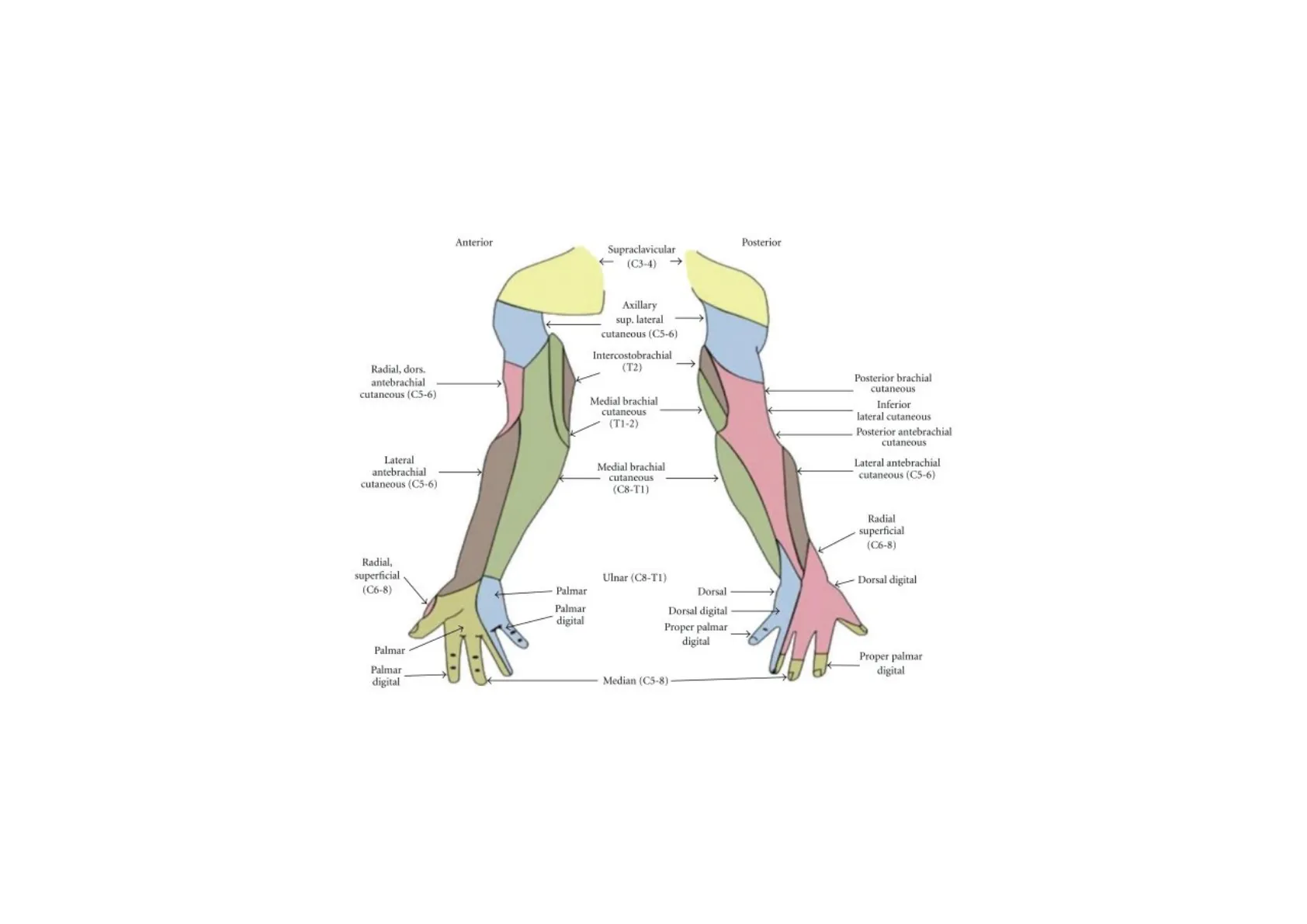
Recuerdo Anatómico
Anterior
Posterior
Supraclavicular
(C3-4) >
Axillary
sup. lateral
cutaneous (C5-6)
Intercostobrachial
(T2)
>
Posterior brachial
cutaneous
Medial brachial
cutaneous
(T1-2)
Inferior
lateral cutaneous
Posterior antebrachial
cutaneous
Lateral
antebrachial
cutaneous (C5-6)
Medial brachial
cutaneous
(C8-T1)
Lateral antebrachial
cutaneous (C5-6)
Radial
superficial
(C6-8)
Radial,
superficial
(C6-8)
Ulnar (C8-T1)
Dorsal digital
Palmar
Dorsal
Palmar
digital
Dorsal digital
Proper palmar
digital
Palmar
Palmar
digital
Median (C5-8)
Proper palmar
digital
Radial, dors,
antebrachial
cutaneous (C5-6)
Recuerdo Anatómico de Nervios
Last thoracic nerve (T12)
Lat. fem. cut. (L2-3)
Lat. cut. branch of
iliohypogastric (L1)
Lumboinguinal, of
genitofemoral (L1-2)
Superior cluneal
(post. div. of L1-3)
Ilioinguinal (L1)
Middle cluneal
(post. div. of S1-3)
Perforating cut.
not colored
Post. fem. cut.
(S1-3)
Cut. obturator
not colored
Anterior
femoral
cutaneous
(L2-3)
Lat. fem. cut
(L2-3)
Lat. fem.
cut. (L2-3)
Com. peroneal
lat. sural cut.
(L4-S2)
Com. peroneal
lat. sural cut.
(L2-3)
Saphenous, medial
crural cut. (L3-4)
Superior peroneal
(L5-S1)
Medial sural cut.
Sural (S1-2)
Sup. peroneal
(L4-51)
Tibial, medial
calcaneal (S1-2)
Lateral dorsal cut.
Lateral calcaneal
Sural, lateral
dorsal cut. (S1-2)
Deep peroneal
(L4-5)
Recuerdo Anatómico de Troncos Nerviosos
C4
C5
Tronco Superior
R.A
C6
. P
Fasciculo Lateral
R.A
Tronco Medio
C7
R. P
N.Axilar
Fasciculo Posterior
R. P
Tronco Inferior
C8
N. Radial
T1
Fasciculo Medial R.A
T2
N. Mediano
N. Cubital
N. MusculoCutaneo
Recuerdo Anatómico de Nervios Lumbosacros
Nervio subcostal ( T12)
T12
Ramos comunicantes
blancos y gris
L1
Nervio iliohipogástrico
L 2
Nervio ilioinguinal
Nervio genitofemoral
L3
Nervio cutáneo lateral
femoral
Ramos comunicantes
L4
gris
N. Musculos pso
e iliaco
L5
Nervio femoral
N. Obturador accesorio
División anterior
División posterior
Nervio obturador
Tronco lumbosacro
Clasificación de Neuropatías
Formas de Clasificación
- SINTOMATOLOGIA PREDOMINANTE
-Motora, sensitiva, autonómica o mixta - TIEMPO DE INSTAURACIÓN
-Aguda (>4 semanas), subaguda (4-8 semanas), crónica (>8 semanas) o recidivante - DISTRIBUCIÓN ANATÓMICA
-Mononeuropatia, mononeuropatia múltiple, polineuropatia, radiculopatía, plexopatía - CARACTERÍSTICAS LESIÓN NERVIOSA (EMG)
-Axonal, desmielinizante - ETIOLÓGICA
Polineuropatías
Clasificación Etiológica de las Polineuropatías
- Hereditarias
-Charcot Marie Tooth o sensitivomotora hereditaria
-Hereditaria por sensibilidad a la presión o tomacular
-Amiloidótica familiar
-Sensitiva y disautonómica hereditaria
-Motora hereditaria o atrofia muscular espinal
-Bulboespinal ligada al cromosoma X - Adquiridas
-Metabólicas, tóxicas y carenciales
-Asociadas a enfermedad sistémica: Neoplasias, vasculitis, paraproteinemias, infecciosas
-Neuropatía del enfermo crítico
-Inmunomediadas
-Por atrapamiento, compresión y otros agentes físicos
Epidemiología de Neuropatías Periféricas
Las neuropatías periféricas tienen una incidencia de 77/100.000
habitantes por año
Y una prevalencia del 1-12% en todas los grupos de edad y hasta un
30% en personas >70años
Aproximadamente un 20% de los casos no se consigue diagnóstico
etiológico
Estudios Epidemiológicos
Los estudios epidemiológicos basados en datos hospitalarios proporcionan
listas (a menudo diferentes) de las causas mas frecuentes de neuropatía
periférica en países occidentales
Table 1 Causes of peripheral neuropathy according to studies in Norway and the Netherlands
Norway [5]
Netherlands [1]
USA [6]
Number of patients:
226
743
231
Idiopathic axonalb
28%
26%
12%
Diabetic
18%
32%
46%
Toxic (alcohol, drugs chemotherapy etc.)
10%
14%
13%
Inflammatory / Immune-mediated
16%
9%
8%
Hereditary
14%
5%
7%
Vasculitic, amyloid neuropathy, sarcoid, connective tissue disease
a
5%
1%
Uremic, thyroid dysfunction
a
4%
3%
Vitamin B12 deficiency
4%
3%
1%
Others (i.e. idiopathic small fiber neuropathyb)
10%
2%
a= not classified, b = axonal in the study from the Netherlands
Polineuropatías Hereditarias
Herencia de Polineuropatías
Padre enfermo
Madre sana
Enfermos
50%
Sanos
50%
Madre portadora
Padre sano
Madre enferma
XY
XX
XY
xx
XY
XX
Sanos
50%
Enfermos
50%
Sano
25%
Portadores
50%
Enfermo
25%
- Variabilidad clínica y genética
- Historia familiar:
-AD
-AR
-Ligada cr. X
Padre portador - Son enferemdades heterogéneas donde la neuropatia puede no ser el principal motivo de consulta
Enfermedad de Charcot Marie Tooth
Es uno de los trastornos neurológicos mas frecuentes (3,6 casos/100.000 hab)
- Neuropatia sensitivo-motora con diferentes subtipos
- El subtipo 1a (60%) y el subtipo 2 (22%) son los mas frecuentes
- Herencia predominante AD
- Mutación mas frecuente (90% de las CMT1a) es la duplicación del gen PMP 22 con locus en cr. 17
-variabilidad familiar debido a penetrancia gen
Manifestaciones de Charcot Marie Tooth
Inicio en la infancia y manifestación más acusada 2ª década
- Predominan síntomas motores:
-atrofia muscular distal eeii (piernas de cigüeña) - Los signos sensitivos aparecen tardiamente y afectan a sensibilidad superficial y profunda (pies
cavos) - Los RMP suelen estar disminuidos o abolidos
- Biopsia de nervio sural: Formaciones 'en bulbo de cebolla '
nervios palpables
Clasificación de CMT
CMT 1
(1A, 1B,
1C, 1D)
CMT 2
CMT 3
CMT 4
CMT X
Desmielinizante
Axonal
AD
AD
AD/AR
AR
Ligada al
cromosoma X
1A: duplicación
gen PMP22 del
cromosoma 17
Mutaciones
puntuales del
gen PO, entre
otros genes, del
cromosoma 1
Duplicación
del gen PMP22
Mutaciones
puntuales
del gen
GDAP1 del
cromosoma 8
Mutaciones
puntuales del
gen la conexina
32 del
cromosoma X
1B: mutaciones
puntuales del
gen PO del
cromosoma 1
Mutaciones
puntuales del
gen PO del
cromosoma 1
AD:AUTOS. DOMINANTE; AR: AUTOS. RECESIVO; PMP22: PROTEÏNA MILÍNICA PERIFÉRICA 22
PO: PROTEÍNA MIELÍNICA 0; GDAP1:PROTEÍNA 1 ASOCIADA A GANGLIÓSIDO
Neuropatía Hereditaria por Sensibilidad a la Presión
- Herencia AD:
-Delección gen PMP 22 cr. 17 - Mononeuropatía aguda recidivante:
-Debilidad o parestesias tras el mantenimiento de una postura no muy forzada - Nervios expuestos a presión:
-Peroneo (CPE), radial, cubital y mediano - Recuperación lenta, con el tiempo posibilidad secuela
- No tratamiento específico, se aconsejan medidas higiénicas
Neuropatía Motora Hereditaria o Atrofia Muscular Espinal del Adulto
- AME infantiles són más graves (tipos I al III)
- AME del adulto (tipo IV)
-> 20 años
-AR (30% AD)
-Atrofia muscular y debilidad lentamente progresiva
-Clínica mm.inferiores y de cinturas
-No afectación respiratoria ni bulbar - Diagnóstico diferencial con otras miopatías sobre todo las de cinturas (EMG, biopsia muscular)
- Más del 90% de los casos de AME presentan deleción del gen SMN1
Polineuropatías Adquiridas
- Metabólicas (DM e I.Renal), Tóxicas y Carenciales
- Asociadas a neoplasias , vasculitis, infecciosas, paraproteinemias
- Del enfermo crítico
- Neuroinmunomoduladas
- Por atrapamiento, compresión y otros agentes físicos
Neuropatía Diabética
> Neuropatia más frecuente en occidente y asociada a cualquier tipo de DM
-Diferentes síndromes
> Clasificación:
- Formas generalizadas: (relación directa metabólica)
-Polineuropatia simétrica distal sensitivo-motora
-Neuropatía sensitiva aguda dolorosa
-Neuropatía autónoma - Formas focales o multifocales: (no tan específica, relacionadas con alteración vascular o
inflamatoria)
-Neuropatías craneales
-Neuropatía motora proximal o amiotrofia diabética (sdr. de Bruns-Garland)
-Multineuropatia múltiple
Forma Simétrica Distal S-M
> Forma simétrica distal s-m.
- Forma mas frecuente
- Prevalencia > 50% a los 5 años de dm
- Inicio progresivo y predominio afectación sensitiva:
-Positiva: parestesias, dolor urente, alodinia
-Negativa: hipoestesia, ataxia sensitiva
Deficit motor mas tardío, leve y predominio distal - Complicaciones tardías: cambios tróficos (úlceras piel) y articulares
- Diagnóstico clínico que se confirma con EMG
Neuropatia sensitiva aguda:
-Síntomas positivos agudos; EMG normal
Neuropatía Autónoma y Motora Proximal
> Neuropatía autónoma:
- Hipotensión ortostática, disfunción eréctil
- Alteración cardiovascular, vaciamiento gástrico, tránsito intestinal
- Diagnóstico diferencial (fármacos, alteraciones hormonales, neoplasias, neuropatías
autónomas hereditarias ... )
> Neuropatia motora proximal o amiotrofia
-Etiología inflamatoria inmunomediada que afecta de forma uni o bilateral a la región
proximal en las EEII
-Plexopatía lumbosacra aguda: dolor agudo, debilidad motora y amiotrofia (EMG, RM)
>
Mononeuropatia múltiple:
-Mecanismo vascular; DD con vasculitis
Neuropatía en la Insuficiencia Renal
- Prevalencia > 70%, paucisintomática
-Gravedad proporcional a grado IR - PNP sensitivo-motora axonal distal
-Signos motores tardíos, no alteración autonómica - Asocia calambres ,piernas inquietas y en ocasiones prurito (casos graves)
- Frecuentemente combina neuropatías por atrapamiento
- Etiologia tóxica; mejora con diálisis
Neuropatías Tóxicas
- Neuropatía alcohólica: (puede asociarse a encefalopatía)
-Consumo oh crónico con malnutrición (efecto tóxico + carencial)
-PNP senstiva distal (1º eeii; 2° eess) axonal (alt. sensitiva global) - Fármacos:
-Generalmente de tipo axonal (vincristina, otros QT)
-Clínica sensitiva distal y posterior asocia alt. motora
-Retirada fármaco: mejoria en función alteración nervio, tiempo exposición - Otros tóxicos:
- Industriales: PNP axonal sensitiva o s-m
- Plomo: PNP motora pura
- Mercurio: PNP sensitiva pura
-Organofosforados: PNP s-m de inicio tardío a la intoxicación
Neuropatías Carenciales
- Déficit vitamina B: neuropatías desmielinizantes (Consumo tóxicos o alt. gastrointestinales)
VIT B12:
-Degeneración combinada medular (cordón posterior) y PNP sensitiva distal
-Puede asociar signos piramidales y déficit cognitivo
> VIT B6:
-Signos sensitivos positivos EEII
-Asociado a fármacos que consumen B6 (Isoniacida)
> VIT B1:
-Similar a neuropatia por OH: parestesias distales y encefalopatía
-Asociada a consumo arroz y alcoholismo crónico
¿Non has encontrado lo que buscabas?
Explora otros temas en la Algor library o crea directamente tus materiales con la IA.
