Principios básicos de farmacocinética: absorción, distribución y metabolismo
Documento de Universidad sobre Principios Basicos de Farmacocinetica. El Pdf explora los mecanismos de paso a través de membranas celulares y los factores que influyen en la absorción, el fenómeno del primer paso y el papel del citocromo P-450 en el metabolismo de los fármacos, útil para estudiantes de Biología.
Ver más12 páginas

Visualiza gratis el PDF completo
Regístrate para acceder al documento completo y transformarlo con la IA.
Vista previa
Farmacocinética
Estudia el curso temporal de las concentraciones y cantidades de los fármacos, y de sus metabolitos, en los líquidos biológicos, tejidos y excreciones, así como su relación con la respuesta farmacológica, y construye modelos adecuados para interpretar estos datos
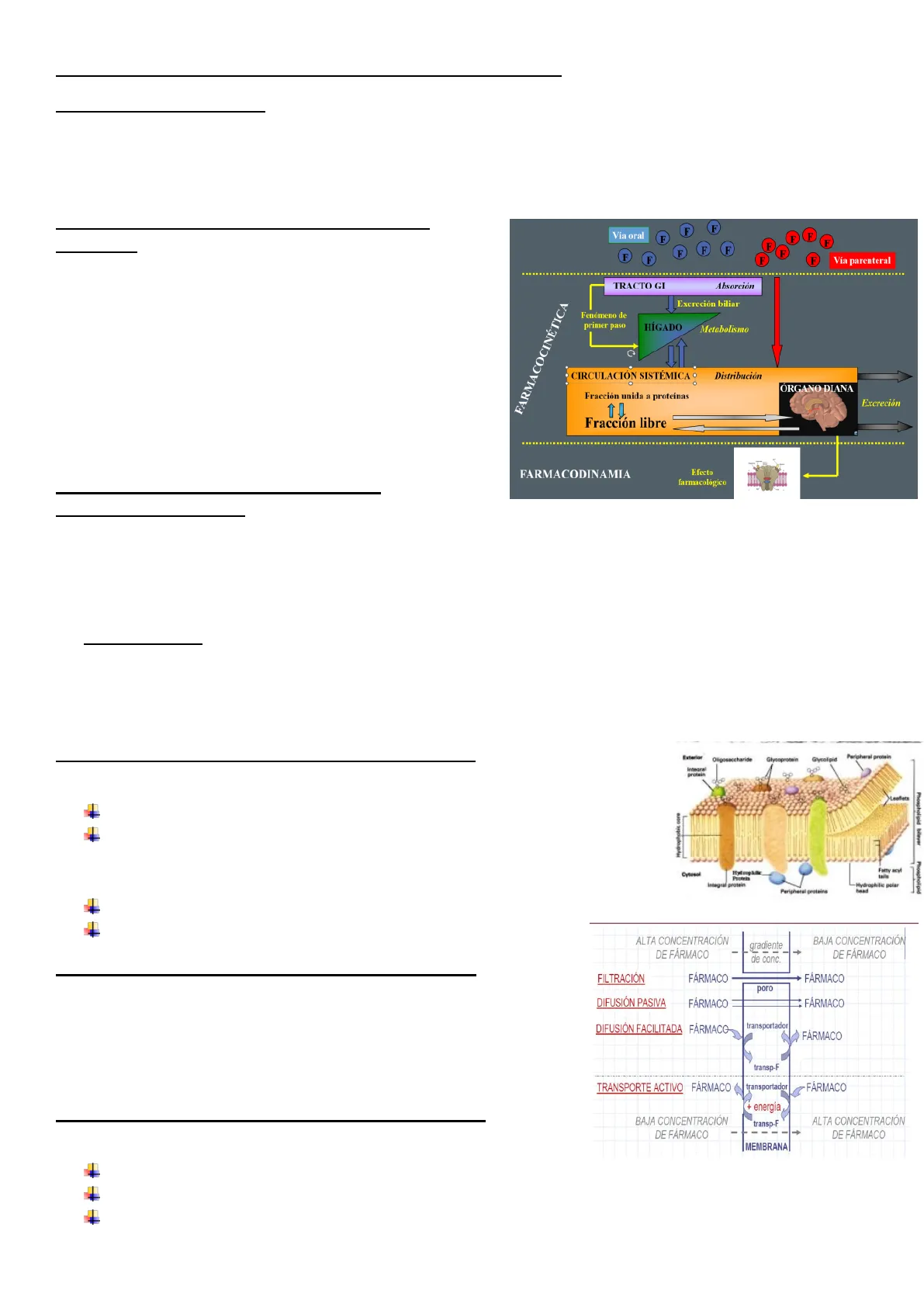
Ciclo intraorgánico del fármaco (LADME)
- Liberación: (de su forma farmacéutica)
- Absorción: disponibilidad sistémica (vías de administración)
- Distribución: liposolubilidad y unión a proteínas plasmáticas
- Metabolismo: biotransformación
- Excreción: eliminación (orina, etc.)
- Eliminación = Metabolismo + Excreción
Importancia del estudio de la farmacocinética
- Comprender los parámetros que definen el curso y el destino del fármaco
- Individualizar el tratamiento farmacológico según las necesidades del paciente
- Monitorizar medicamentos con un índice terapéutico estrecho
- Disminuir el riesgo de efectos adversos y optimizar la respuesta terapéutica
Absorción
Comprende las etapas de liberación del fármaco de su forma farmacéutica, su disolución, la entrada al organismo desde el lugar de administración, los mecanismos de transporte, la velocidad y la cantidad de fármaco que accede a la circulación sistémica.
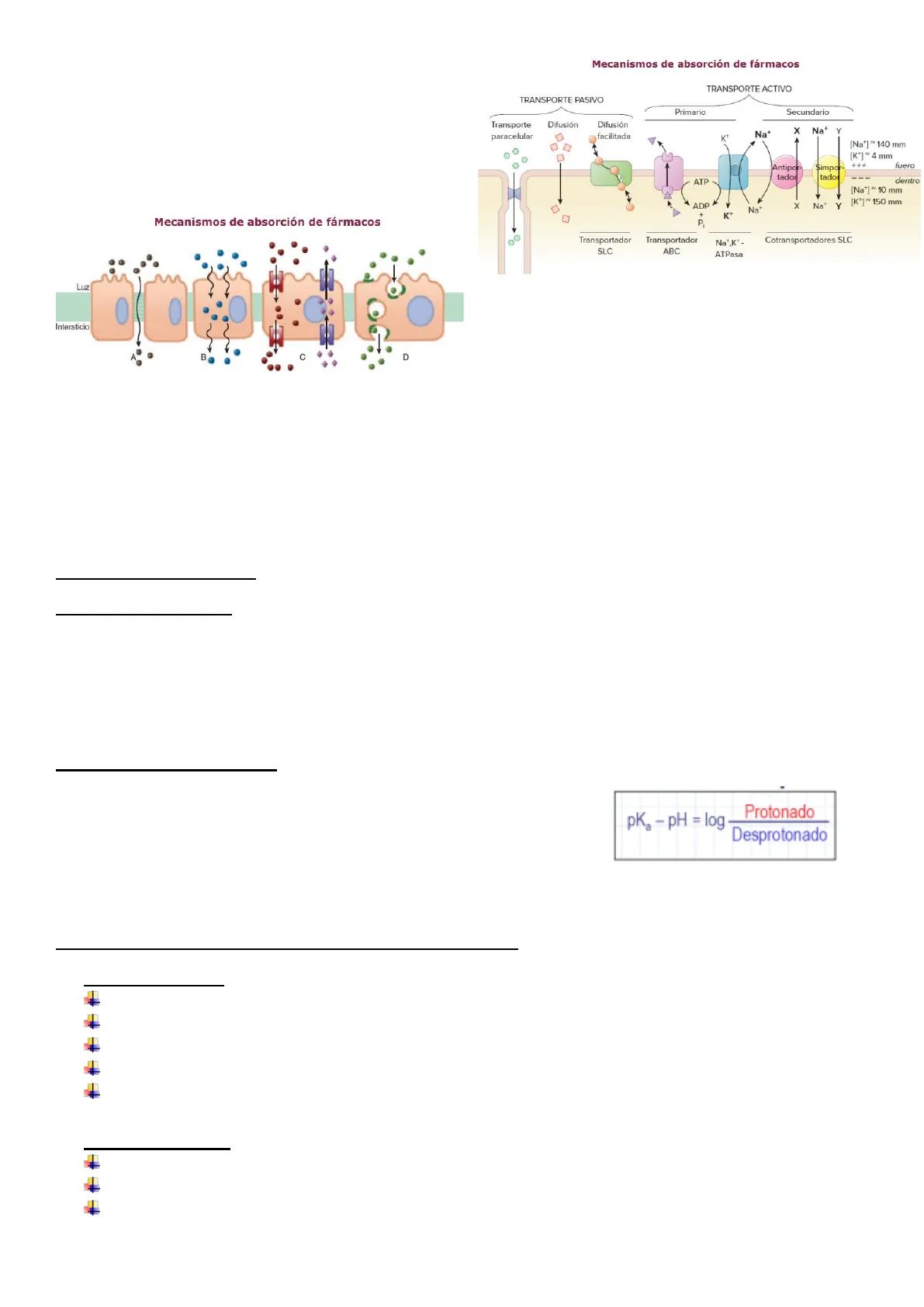
Paso a través de membranas celulares
- La membrana celular es una bicapa lipídica:
- Cabezas polares hacia el exterior
- Cadenas HC hacia el interior
- Incluye proteínas de transmembrana (receptores, canales, transportadores)
- La membrana celular es permeable a:
- Compuestos lipofílicos
- Agua, alcohol,
Mecanismos de absorción de fármacos
- Difusión pasiva / osmosis.
- Difusión facilitada.
- Transporte activo.
- Endocitosis / Pinocitosis
Factores que influyen en la absorción de los fármacos
- Las características fisicoquímicas del fármaco:
- Liposolubilidad (log Po/w)
- Grado de ionización, dependiente de su pKa y del pH del medio
- Tamaño molecular (peso molecular < 1000: la mayoría de los fármacos)
Exterior Digoseccharide Glycoprotein Glycolipid Integra protein >Leeffets Phospholipid Braver Hylsophilk Fatty acyl tall Integral protein Peripheral proteins Hydrophilic polar head ALTA CONCENTRACIÓN DE FÁRMACO gradiente BAJA CONCENTRACIÓN DE FÁRMACO de conc. FILTRACIÓN FÁRMACO FÁRMACO DIFUSIÓN PASIVA FÁRMACO FÁRMACO DIFUSIÓN FACILITADA FÁRMACO transportador FÁRMACO transp-F TRANSPORTE ACTIVO FARMACO transportador FÁRMACO + energía BAJA CONCENTRACIÓN DE FÁRMACO transp-F ALTA CONCENTRACIÓN DE FÁRMACO MEMBRANA F F F F F F Vía parenteral TRACTO GI Absorción Excreción biliar FARMACOCINÉTICA Fenómeno de primer paso HÍGADO Metabolismo CIRCULACIÓN SISTÉMICA · Distribución ÓRGANO DIANA Fracción unida a proteínas Excreción Fracción libre FARMACODINAMIA Efecto farmacológico F F Vía oral F F F F F F F Peripheral protein Cytosol Phospholipid poro· Las características y tipo de la preparación farmacéutica · Las características del lugar de absorción (flujo sanguíneo, etc.) · La eliminación presistémica y el fenómeno de "primer paso" Mecanismos de absorción de fármacos Luz Intersticio B D Los fármacos pueden difundirse pasivamente por conductos acuosos en las uniones intercelulares (p. ej., zonas de oclusión, A) o a través de las membranas lipídicas (B). Los fármacos con las características apropiadas son llevados al interior o exterior de las células por transportadores (C). Los fármacos con dificultades para pasar a través de la membrana pueden unirse con receptores en la superficie celular (sitios de unión de color oscuro), ser circundados por la membrana (endocitosis) y luego liberados dentro de la célula, o expulsados en vesículas delimitadas por membrana fuera de la célula hacia el espacio extracelular (exocitosis, D).
Transporte pasivo
Liposolubilidad
Los fármacos liposolubles atraviesan sin dificultad las membranas celulares de forma pasiva, mediante gradiente de concentración Coeficiente de reparto Lípido / Agua: Log Po/w (octanol/agua):
- Alto: fármacos liposolubles (anestésicos generales)
- Bajo: fármacos hidrosolubles (la mayoría)
Grado de ionización
Muchos fármacos hidrosolubles pueden atravesar pasivamente las membranas celulares al ser electrolitos (ácidos o bases) débiles si:
- Son menores de 100 Dalton
- Se encuentran en forma no ionizada
pKa = pH al que el 50% de las moléculas de un fármaco se encuentran no ionizadas. Para determinar la tasa de las 2 formas se puede usar la ecuación de Henderson-Hasselbalch.
Transporte mediado por transportadores
- Transporte activo
- Funciona contra el gradiente electroquímico y requiere energía.
- Saturable
- Selectivo: susceptible de inhibición competitiva
- Inhibido por mecanismos o substancias que interfieran con la producción de energía.
- Se ha observado en: tubo digestivo, túbulo renal, árbol biliar, en el paso del LCR a sangre y en el paso de la sangre a la glándula salival.
- Difusión facilitada
- A favor del gradiente de concentración, mediante proteínas transportadoras y sin precisar energía
- Saturable
- Selectivo
Mecanismos de absorción de fármacos
TRANSPORTE ACTIVO TRANSPORTE PASIVO Primario Secundario Transporte paracelular Difusión Difusión x Na+ Y facilitada K+ O O [Na+]~ 140 mm [K+]~ 4 mm +++ Antipor- $impor- fuera tador tador ATP dentro ADP > X Na+ Y K P Transportador SLC Transportador ABC Na+,K+- ATPasa [Na]]~ 10 mm [K*]~ 150 mm Na + Cotransportadores SLC . pKa - PH = log Protonado Desprotonado Na+ABSORCION:
Principales transportadores de fármacos
Glicoproteína P (Pgp)
Función: Excreción de xenobióticos y metabolitos a orina o bilis, y luz intestinal y prevención de su acumulación en cerebro ("herramienta de defensa de los territorios nobles del organismo").
- (P = "permeabilidad")
- Proteína bomba de flujo de salida (dependiente de energía) de gran tamaño (170kDa) que exporta sus substratos al exterior de la célula
Biodisponibilidad
Fracción de la dosis administrada que llega inalterada al torrente circulatorio. Depende básicamente de la absorción, pero también de la distribución y eliminación. Depende de:
- Vía de administración (100% i.v.)
- Forma farmacéutica
- Posibles interacciones con otros fármacos o alimentos
- Degradación en el estómago, metabolización hepática o intestinal ...: "efecto de primer paso" o
eliminación presistémica
- Fenómeno de primer paso Se refiere a la metabolización hepática que se produce del fármaco absorbido en el tracto gastrointestinal y que llega al hígado por la vena porta antes de entrar en la circulación sistémica
- Se representa con la letra F y se expresa en porcentaje o con una fracción de 0-1. Ejemplo: F = 0,6 ó 60%.
- Relación entre el curso temporal de la concentración plasmática de un fármaco en el organismo y sus efectos.
- CMT = concentración máxima tolerada Aquella por encima de la cual suelen observarse efectos tóxicos.
- CME = concentración mínima eficaz Aquella por encima de la cual se observa el efecto terapéutico
- AUC (ABC) = área bajo la curva Concentración del fármaco en el plasma en función del tiempo
- PL = periodo de latencia Tiempo que transcurre desde la administración hasta el inicio del efecto (hasta que la concentración plasmática alcanza la CME)
- TE = tiempo eficaz (duración del efecto) Tiempo transcurrido entre que se alcanza la CME y el momento en que desciende por debajo de ésta.
- IE = Intensidad del efecto Depende (para muchos fármacos) de la concentración máxima que se alcance
Influencia de las vías de administración y de las formas farmacéuticas sobre las curvas de concentraciones plasmáticas de un fármaco
Vías de administración
Concentración plasmática IV IM Oral Rectal Saber representar las gráficas Formas farmacéuticas: Tiempo Forma farmaceutica 2 Concentración plasmática Solución oral Cápsulas Comprimidos
Distribución
- Transporte del fármaco (sistema sanguíneo y linfático) a los órganos en los que debe actuar y a los órganos que lo van a eliminar y condiciona las concentraciones que alcanzan en cada tejido.
- Las moléculas de un fármaco son transportadas en la sangre disueltas en el plasma, fijadas a proteínas plasmáticas o unidas a células sanguíneas.
- Factores que determinan la distribución de los fármacos
- Propiedades físico-químicas del fármaco
- Fijación a proteínas del plasma
- Fijación a proteínas tisulares
- Perfusión sanguínea tisular
Transporte plasmático - Unión a proteínas plasmáticas
- De la unión a proteínas plasmáticas, la fijación a albúmina es la más frecuente e importante.
- La al-glicoproteína ácida es otra proteína importante para la unión, además de las lipoproteínas.
- Tras su ingreso en el torrente sanguíneo, una parte del fármaco de une a proteínas plasmáticas
(reservorio inactivo) y otra porción circula libre (fracción activa), disponible para atravesar las
membranas biológicas (unirse a receptores, distribuirse por tejidos corporales, ser sometida a
metabolismo y excreción).
- La unión F+PP es reversible (equilibrio asociación-disociación) y saturable
- Existe la posibilidad de desplazamiento debido a competición entre F por los lugares de unión (también con los ligandos endógenos).
Unión de diferentes fármacos a la albúmina plasmática
- Fármacos liposolubles: acceso fácil a órganos irrigados (cerebro, corazón, hígado, riñones).
- Si un fármaco A unido en gran proporción a seroalbúmina es desplazado por otro, B, que actúa como competidor, la disponibilidad de A "libre" (no unido) aumenta rápidamente, lo que puede conducir a una exacerbación de su efecto (interacción farmaco- fármaco)
- El desplazamiento por fármacos (e.g. sulfonamidas) de la bilirrubina no conjugada unida a seroalbúmina puede precipitar una encefalopatía por bilirrubina ("kernícterus") en el recién nacido.
- Fármacos menos liposolubles: acceso fácil a tejidos cuyos capilares son ricos en hendiduras intercelulares (sinusoides hepáticos).
- La mayoría de los fármacos tienen la capacidad de fijarse a determinados tejidos en los que alcanzan concentraciones más altas que en el resto del organismo. Ej .: acumulación de fármacos liposolubles en la grasa.
Perfusión sanguínea
- La magnitud del flujo sanguíneo determina la velocidad de distribución de los fármacos y en
consecuencia produce
- Altas concentraciones en órganos bien perfundidos
- Bajas concentraciones o lenta acumulación en los mal perfundidos
Reservorios tisulares de fármacos
Tejido adiposo
- Fármacos liposolubles:
- Barbitúricos
- Beta-bloqueantes adrenérgicos
- Reservorio estable, Bajo flujo sanguíneo
- Obesidad
¿Non has encontrado lo que buscabas?
Explora otros temas en la Algor library o crea directamente tus materiales con la IA.
