Patología de la Coagulación Intrínseca y la Fibrinólisis, Apuntes
Documento de Universidad Miguel Hernández sobre Patología de la Coagulación Intrínseca y la Fibrinólisis. El Pdf detalla las fases de la hemostasia, los mecanismos de anticoagulación y fibrinólisis, pruebas de laboratorio y un algoritmo de diagnóstico diferencial para diátesis hemorrágicas en Biología.
Ver más13 páginas


Visualiza gratis el PDF completo
Regístrate para acceder al documento completo y transformarlo con la IA.
Vista previa
Patología de la Coagulación Intrínseca y la Fibrinólisis
Francisco López García Patología General Patología de la coagulación intrínseca y la fibrinólisis Clase Nº30 - (05/05/23) PG
Contenido de la Clase
PATOLOGÍA DE LA COAGULACIÓN INTRÍNSECA Y LA FIBRINÓLISIS CONTENIDO
- VÍA INTRÍNSECA DE LA COAGULACIÓN
- FASES DE LA HEMOSTASIA
- VÍA INTRÍNSECA
- PRUEBAS DE LABORATORIO
- PATOLOGÍA DE LA COAGULACIÓN INTRÍNSECA
- MECANISMO MOLECULAR DE LA HEMOFILIA Y DE LA ENFERMEDAD DE VON WILLEBRAND
- MANIFESTACIONES CLÍNICAS
- FIBRINÓLISIS
- MECANISMOS DE ANTICOAGULACIÓN
- INHIBIDORES DE LA FIBRINOLISIS
- PATOLOGÍAS
- PRUEBAS DE LABORATORIO
- COAGULACIÓN INTRAVASCULAR DISEMINADA
- PATOGENIA
- ETIOLOGÍA
- MANIFESTACIONES CLÍNICAS
- PRUEBAS DE LABORATORIO
- DIÁTESIS HEMORRAGICA-ALGORITMO DE DIAGNÓSTICO DIFERENCIAL
Lucía Muela Muela Universidad Miguel Hernández. Segundo - Grado Medicina 2022/2023 Medicumh @ Página 1 de 11Francisco López García Patología General Patología de la coagulación intrínseca y la fibrinólisis Clase Nº30 - (05/05/23) PG
Vía Intrínseca de la Coagulación
En la clase anterior se explico la patología de la vía extrínseca de la hemostasia. Esta comisión trata de la patología de la vía intrínseca y de la fibrinólisis.
Fases de la Hemostasia
- Hemostasia primaria (provisional): mecanismo inicial para detener la hemorragia al lesionarse el vaso. Intervienen las plaquetas y los vasos sanguíneos. El resultado final es la formación del trombo plaquetario. En la mayoría de los casos no detiene totalmente la hemorragia.
- Hemostasia secundaria (definitivas): se produce cuando se activa a modo de cascada de factores de coagulación o proteínas plasmáticas con el objetivo, gracias a la acción de plaquetas, fibrinogeno y hematíes, de formar coágulos de fibrina. Presenta dos vías de activación, la extrínseca y la intrínseca. Nos centraremos en la vía intrínseca para entender después su patología.
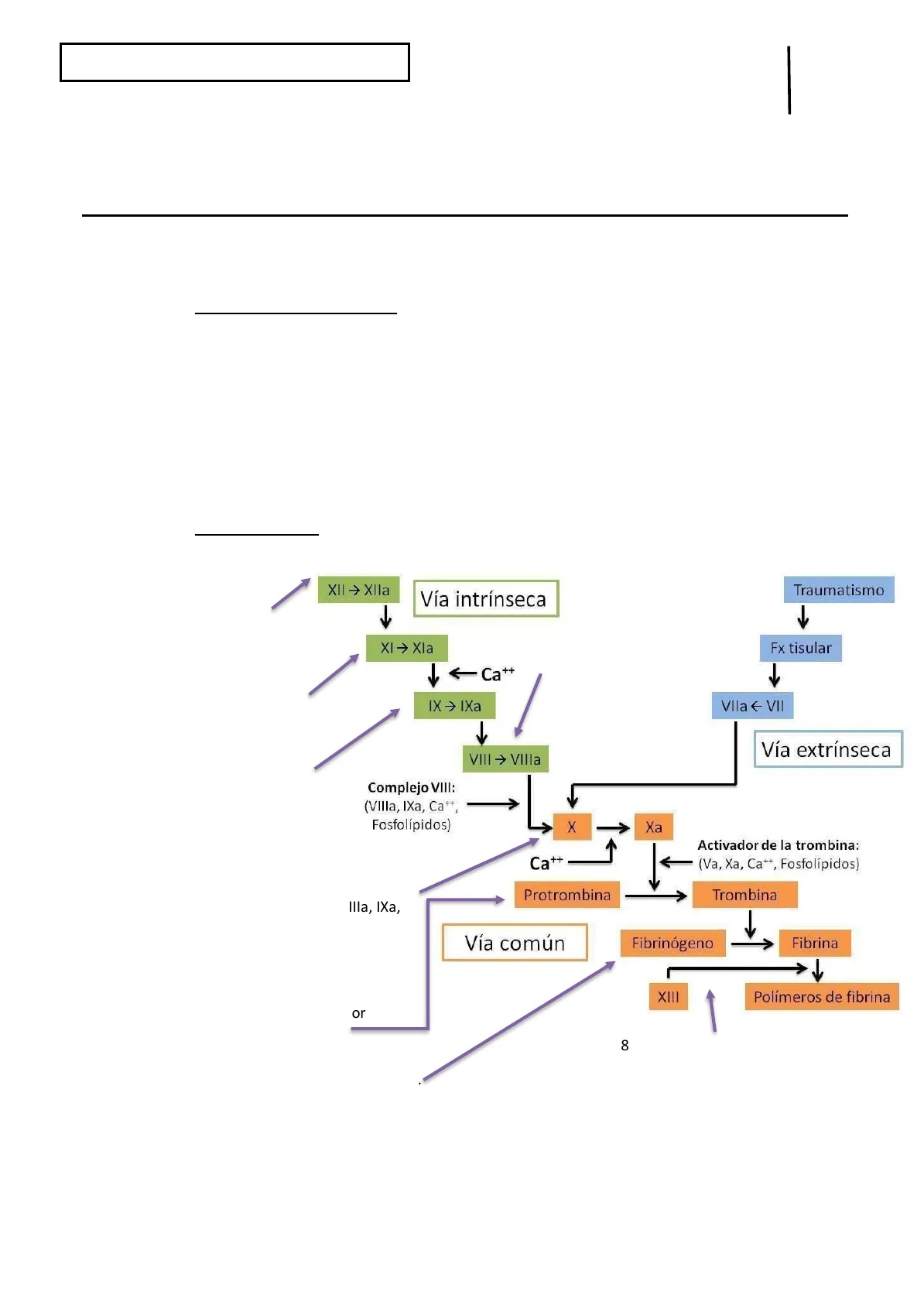
Vía Intrínseca
- Activación del factor XII Traumatismo (XII > Xlla) debido al contacto de la sangre con la membrana basal del endotelio y con superficies polianiónicas. XII -> XIla Vía intrínseca XI -> Xla Fx tisular Ca+
- Activación del factor XI (XI > Xla) por acción del factor XIIa. VIII > Villa 3. Activación del factor IX (IX -> IXa) por acción del facto XIa y la intervención deCa2+. Complejo VIII: (VIIla, IXa, Ca++, Fosfolípidos) X Xa Ca++ Activador de la trombina: (Va, Xa, Ca ** , Fosfolípidos) Protrombina Trombina Vía común Fibrinógeno Fibrina 7. Conversión de fibrinogeno a fibrina por la trombina. 8. Formación del coágulo de fibrina debido a la previa conversión del fibrinogeno en fibrina y a su polimerización por el factor XIII.
Las vías extrínseca e intrínseca se comunican en la activación del factor X, por lo que a partir de este momento se considera vía final común.
Lucía Muela Muela Universidad Miguel Hernández. Segundo - Grado Medicina 2022/2023 Medicumh @ Página 2 de 11 5. Activación del factor X (X-> Xa) por participación del complejo VIII donde están incluidos VIIIa, IXa, Ca2+ y fosfolípidos. 6. Conversión de protrombina a trombina debido a la actuación del complejo activador de la trombina: Va, Xa,Ca2+ y fosfolípidos. XIII Polímeros de fibrina IX -> IXa Vía extrínsecaFrancisco López García Patología General Patología de la coagulación intrínseca y la fibrinólisis Clase Nº30 - (05/05/23) PG
La vía intrínseca de la coagulación sanguínea se produce cuando la sangre entra en contacto con ciertas sustancias, como las endotoxinas bacterianas, o por otros mecanismos que pueden desencadenar la secuencia enzimática al activar un tipo de factores llamados proteínas de contacto. La activación de la vía intrínseca y extrínseca contribuye a la formación del complejo protrombinasa, que se sigue de la activación de la "vía final común". Con esta vía se llega a la formación de trombina y, luego, de fibrina (ésta última ve favorecida su estabilización por el FXIII).
Pruebas de Laboratorio
Existen diversas pruebas de laboratorio para valorar la vía extrínseca, la intrínseca y la final que es común.
- VÍA EXTRÍNSECA:
- Tiempo de protrombina o índice de Quick (TP): Se alarga por déficit de los factores II (protrombina), VII, X Y V. Ya comentado en la comisión anterior.
- VÍA INTRÍNSECA:
- Tiempo de tromboplastina parcial activado (APTT): Tiempo que tarda en coagular una muestra de plasma después de añadirle un activador de la fase de contacto que inicie la coagulación intrínseca, como el caolín (activador del factor XII), en presencia de calcio y fosfolípidos (cefálica que sustituye a los fosfolípidos plaquetarios). Consiste en realizar in vitro lo que sucede in vivo. Se alarga por déficit de los factores II, VII, IX, XII, X, V. Sirve para evaluar tanto la vía intrínseca como la final común de la hemostasia secundaria. Si los resultados de APTT son normales no se puede excluir deficit de los factores VII y XIII ya que pertenecen a la vía extrínseca.
- Valoración de factores aislados: son pruebas más complejas de mezcla que nos permiten diferenciar déficits de factores de coagulación de la presencia de sustancias con efecto anticoagulante.
- Tiempo de Trombina (TT): valora la vía final comun. Se añade trombina (transforma el fibrinogeno en fibrina) directamente al plasma. Mide el tiempo de coagulación que tarda en coagular el plasma la adición de trombina. Su prolongación refleja anomalías del fibrinogeno, sustancias que impiden la acción de la trombina sobre su substrato (antitrombinas) o trastornos en la polimerización de la fibrina (heparina). Por lo tanto, el alargamiento del TT se debe principalmente a una cantidad baja de fibrinogeno o alta de antitrombina.
LA VÍA INTRÍNSECA DE LA COAGULACIÓN EL APTT 双 双 以 男 VB + FT Via intrinseca Via extrinseca TTPA TP x Via común Medicumh@ Página 3 de 12 Lucía Muela Muela Universidad Miguel Hernández. Segundo - Grado Medicina 2022/ Protrombina Trombina TFrancisco López García Patología General Patología de la coagulación intrínseca y la fibrinólisis Clase Nº30 - (05/05/23) PG
Patología de la Coagulación Intrínseca
La patología de la coagulación intrínseca corresponde con enfermedades congénitas o adquiridas que afectan a uno o a varios factores de la vía intrínseca, alterando la coagulación plasmática. Pueden deberse a:
- Déficit de síntesis de los factores de coagulación (factores de la vía intrínseca):
- Congénitas:
- Hemofilias A (déficit de factor VIII coagulante) y B (déficit de factor IX ß-coagulante).
- Déficits aislados de factor XI y XII. Estos son menos frecuentes que las hemofilias.
- Adquiridas:
- Insuficiencia hepatocelular.
- Déficit de vitamina K por uso de acenocumarol (sintrom) o colestasis prolongadas.
- Determina deficiencia de factores II (protrombina), VII, IX y X.
- Congénitas:
- Inhibidores de factores de coagulación, como la heparina o el rivaroxaban (nuevo anticoagulante).
. Anticoagulantes circulantes. Estos son anticuerpos dirigidos contra factores de la
coagulación. Entre ellos destaca el anticuerpo anti factor VIII. Puede aparecer en:
- Sujetos santos.
- Pacientes hemofílicos en tratamiento sustitutivo.
- Embarazo.
- Enfermedades autoinmunes.
Mecanismo Molecular de la Hemofilia y de la Enfermedad de Von Willebrand
Normal Hemofilia A Enfermedad de von Willebrand Cromosoma X Cromosoma 12 Cromosoma X Cromosoma 12 Cromosoma X Cromosoma 12 FVIII-C VWF VWF Complejo FVIII-C/ vWF Rápida destrucción de la molécula FVIII-C en ausencia de vWF, que normalmente lo protege
Lucía Muela Muela Universidad Miguel Hernández. Segundo - Grado Medicina 2022/2023 Medicumh@ Página 4 de 12 FVIII-CFrancisco López García Patología General Patología de la coagulación intrínseca y la fibrinólisis Clase Nº30 - (05/05/23) PG
La diapositiva muestra las bases moleculares de la hemofilia A y de la enfermedad de von Willebrand. En el FVIII hay una molécula con actividad coagulante, que es el FVIII:C, codificado por el cromosoma X, y el factor de von Willebrand (vwF), codificado por el cromosoma 12 cuya labor es transportar el factor VIII:C, regulando su aclaramiento. En la hemofilia es defectuosa la síntesis del FVIII:C, mientras que en la enfermedad de von Willebrand no se produce el vwF, con lo que se destruye de forma rápida el FVIII:C
Enfermedad de Von Willebrand
Esta enfermedad es el trastorno hereditario más común de la hemostasia (sus dos vías están afectadas). Constituye un defecto cualitativo o bien cuantitativo del factor de vW. Su patogenia refleja la afectación tanto de la hemostasia primaria como de la secundaria. En la hemostasia primaria, el factor vW interviene en la adhesión plaquetaria; por lo tanto, un defecto de este factor puede provocar hemorragias cutáneomucosas y un alargamiento del tiempo de hemorragia. En la hemostasia secundaria, como ya hemos mencionado, forma un complejo con el factor VIII, por lo cual, por su deficiencia, se van a producir de forma secundaria hematomas y alargamiento del APTT.
Manifestaciones Clínicas
- A menudo son asintomáticos por lo que su hallazgo es casual. Puede deberse a que existe cierta cantidad de factor VIII y de factor de von Willebrand.
- Es un tipo de diátesis hemorrágica plasmopática, provocada por una disminución en el número de plaquetas.
- La hemorragia es diferida, aparece en horas/días tras el traumatismo.
- Aparecen hematomas en heridas quirúrgicas.
- Predominan grandes equimosis y hematomas profundos, cuando nos encontramos ante grandes traumatismos.
- No se producen petequias: la fase inicial (plaquetar) de la coagulación esta intacta.
- Sangrado en tejidos profundos: hematomas musculares, vísceras, retroperitoneo y articulaciones, sobre todo en hemofílicos.
HEMARTROS AGUDO EN LA HEMOFILIA HEMATOMA MASIVO EN EL BRAZO DE UN HEMOFILICO
Lucía Muela Muela Universidad Miguel Hernández. Segundo - Grado Medicina 2022/2023 Medicumh@ Página 5 de 12Francisco López García Patología General Patología de la coagulación intrínseca y la fibrinólisis Clase Nº30 - (05/05/23) PG
GENU RECURVATUM HEMARTROS RECIDIVANTE HEMORRAGIA MASIVA TRAS EXTRACCION DENTAL, FRECUENTE EN LA ENF DE VON WILLEBRAND A ARTRITIS CRONICA HEMARTROS RECIDIV
Fibrinólisis
Mecanismos de Anticoagulación
Los mecanismos de anticoagulación permiten que la fluidez de la sangre se mantenga al inhibirse la hemostasia, impidiendo la coagulación excesiva y/o la disolución del coagulo. Los mecanismos implicados son:
- Inhibidores plasmáticos de la hemostasia:
- Antitrombina III: inactiva la trombina y los factores IX, X, XI y XII de la coagulación mediante una reacciónque se acelera en presencia de heparán sulfato producido por elendotelio.
- Sistema Trombomodulina-Proteina C-Proteina S: el endotelio produce trombomodulina y realiza una doble acción; se une a la trombinainhibiendo su efecto y, el propio complejo trombomodulina- trombina, activa a la proteína C. Esta proteína C activada tiene otra doble acción: inactiva los factores V y VIII e induce la fibrinolisis. La proteína S actúa como cofactor de la proteína C, y recordar que tanto la proteína C como la S dependen de la vitamina K.
Proteína S Inactivación de factores V y VIII Proteína C Proteína C activada Inducción de fibrinólisis Coagulación Trombina Trombomodulina Endotelio
Lucía Muela Muela Universidad Miguel Hernández. Segundo - Grado Medicina 2022/2023 Medicumh @ Página 6 de 12
¿Non has encontrado lo que buscabas?
Explora otros temas en la Algor library o crea directamente tus materiales con la IA.
