Apuntes sobre patologías hematológicas, hemostasia y coagulación
Documento de Universidad sobre Contenido. El Pdf aborda patologías hematológicas como hemocromatosis, mielofibrosis, hemostasia y coagulación, ideal para estudiantes de Biología a nivel universitario.
Ver más26 páginas


Visualiza gratis el PDF completo
Regístrate para acceder al documento completo y transformarlo con la IA.
Vista previa
CONTENIDO
PARCIAL 1
- Hematopoyesis
- Granulopoyesis
- Monopoyesis
- Plaquetas
- Grupo Sanguineos RH
- Citometría hemática
- Anemias
- Anemia Ferropénica
- Carenciales megaloblásticas
- Talasemias
- Esferocitosis Hereditarias
- Anemia Hemolítica
- Autoinmune
- Incompatibilidad de grupos
- Anemias por enf. crónicas
PARCIAL 2
- Hemocromatosis
- Neutropenia
- Trombocitopenia
- Leucemia linfoblástica aguda +
- Leucemia mieloblástica aguda
- Sx linfoproliferativo
- Linfoma NO Hodgkin
- Linfoma Hodking
- Leucemia linfocítica crónica
- Sx mielodisplásico
- Sx mieloproliferativo
- Leucemia Mieloide Crónica
- Mieloma Multiple
HEMOCROMATOSIS
Trastorno del metabolismo del hierro -> Absorción intestinal excesiva y depósito progresivo de hierro en diversos órganos -> Hígado, corazón, páncreas y piel -> Daño tisular y disfunción orgánica.
CAUSAS
- Primaria: Mutaciones en el gen HFE (C282Y y
H63D) -> Alteran la regulación de la absorción
del hierro.
- Otros: HJV, HAMP, TFR2 y SLC40A1 (Ferroportina).
- Secundarias: Transfusiones repetidas, enf hematológicas crónicas (Talasemias, anemias sideroblásticas o Sx mielodisplásicas), enf hepática crónica, consumo excesivo de hierro.
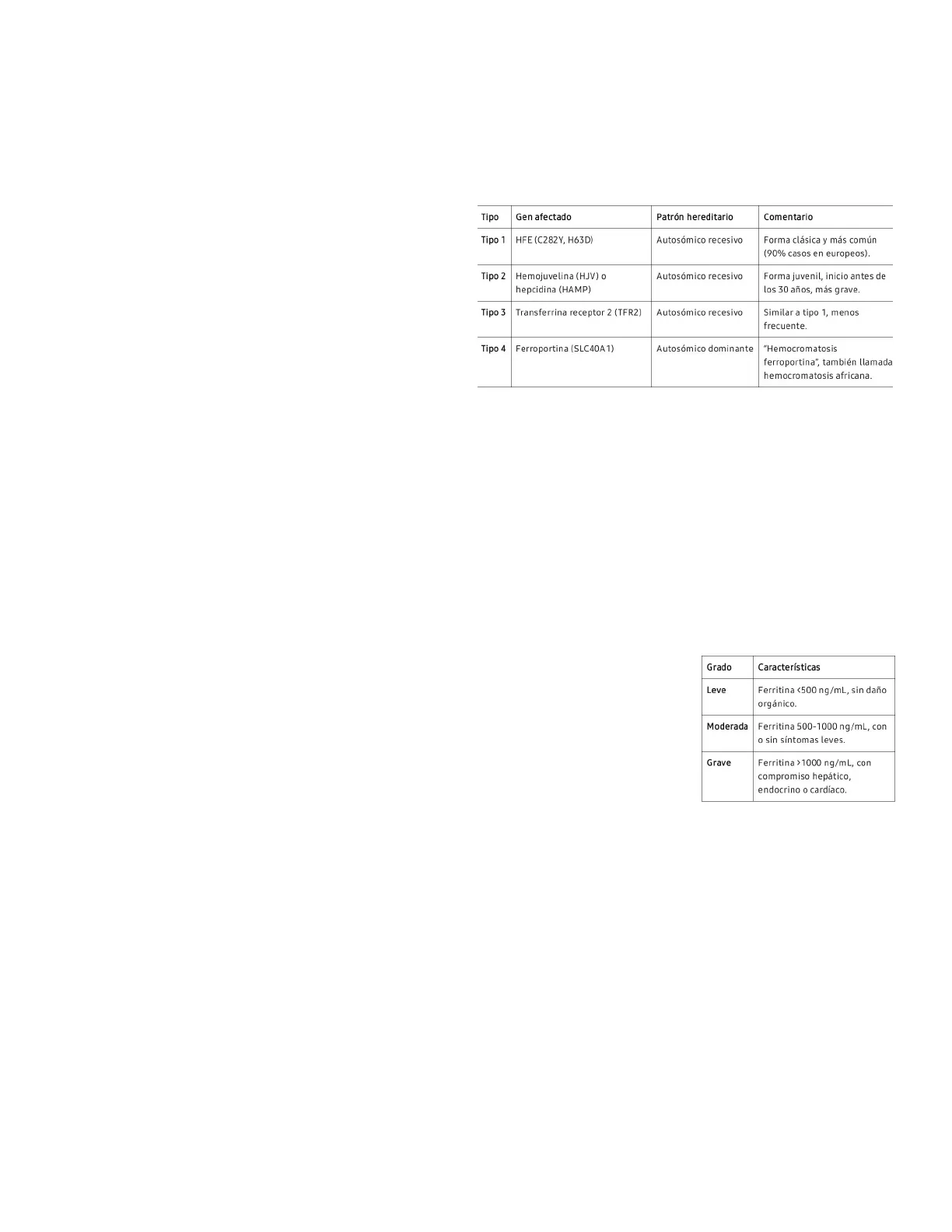
| Tipo | Gen afectado | Patrón hereditario | Comentario |
| Tipo 1 | HFE (C282Y, H63D) | Autosómico recesivo | Forma clásica y más común (90% casos en europeos). |
| Tipo 2 | Hemojuvelina (HJV) o hepcidina (HAMP) | Autosómico recesivo | Forma juvenil, inicio antes de los 30 años, más grave. |
| Tipo 3 | Transferrina receptor 2 (TFR2) | Autosómico recesivo | Similar a tipo 1, menos frecuente. |
| Tipo 4 | Ferroportina (SLC40A1) | Autosómico dominante | "Hemocromatosis ferroportina", también llamada hemocromatosis africana. |
FISIOPATOGENIA
Producción reducida de hepcidina (hormona hepática que regula la absorción intestinal de hierro) -> Aumento de la absorción intestinal de hierro / Liberación sin restricción de hierro desde los enterocitos y macrófagos / Exceso de hierro circulante que se deposita en tejidos -> Promoviendo daño oxidativo y fibrosis.
SÍNTOMAS
Asintomaticos (Inicio): Fatiga crónica / Dolor articular (Manos) / Pigmentación cutánea / DM / Hepatomegalia / Cirrosis / Disfunción cardíaca / Hipogonadismo / Perdida de líbido o impotencia.
GUIA
- Tamizaje en familiares de 1º grado en px con HH.
- Uso de:
- Saturación de transferrina >45%
- Ferritina -> H: >300 ng/ml / M: >200 ng/mL.
- Tto con flebotomía -> Ferritina elevada, aún con ausencia de síntomas.
CLASIFICACIÓN
Severidad (Grado de sobrecarga férrica y daño orgánico) -> Para la clínica.
| Grado | Características |
| Leve | Ferritina <500 ng/ml, sin daño orgánico. |
| Moderada | Ferritina 500-1000 ng/ml, con o sin síntomas leves. |
| Grave | Ferritina > 1000 ng/mL, con compromiso hepático, endocrino o cardíaco. |
DIAGNÓSTICO
- Inicio -> Saturación de transferrina >45% / Ferritina sérica elevada.
- Confirmación -> Estudios genético (Mutación HFE) / RM para cuantificación del hierro hepático / Biopsia hepatica, para estimar el daño y cuantificar el hierro tisular.
- Otros -> Elevación de transaminasas / DM o hipogonadismo en laboratorios hormonales.
HISTOLOGÍA
Acuúmulo de hemosiderina dentro de hepatocitos (o en cél de Kupffer) / Patrón de depósito periportal / Fibrosis progresiva que puede evolucionar a cirrosis / Técnicas especiales -> Tinción con azul de Prusia para hierro.
TRATAMIENTO
- Flebotomías periódicas (Extracción de sangre de 400-500 ml c/1-2 sems, hasta normalizar la ferritina)
- Objetivo -> Ferritina <50-100 ng/ml / Mantenimiento: Flebotomías menos frecuentes.
- Quelantes de hierro (Desferoxamina, deferasirox) -> No se pueda hacer flebotomía
- Modificaciones en la dieta -> Evitar suplementos de hierro, alcohol y vitamina C en exceso.
NEUTROPENIA
Disminución del número absoluto de neutrófilos (NAN) en sangre periférica.
- Leve: 1000-1500/uL
- Grave: < 500/uL
- Moderada: 500-1000/uL
- Muy grave: < 200/uL
Valores de referencia según la edad:
- Niños 1mes - 10a: < 1.5 x 10>9/L.
- >10a: < 1.8 × 109/L.
- Riesgo critico: < 0.5 x 109/L.
CAUSAS
- Congénitas: Sx de Kostmann / Neutropenia cíclica / Trastornos genéticos > Mutaciones en ELANE, HAX1, G6PC3.
- Adquiridas: Fármacos / Infecciones / Autoinmunes / Hipoplasia medular / Hiperesplenismo / Déficits nutricionales.
FISIOPATOGENIA
- Neutropoyesis hipoplásica -> Disminución de producción en médula ósea.
- Destrucción periférica.
- Redistribución o secuestro.
- Neutropoyesis ineficaz -> Alteración en la maduración.
- Aumento de apoptosis
- Infiltración medular o fibrosis
CLÍNICA
- Asintomáticos (Neutropenia leve)
- Grave: Fiebre persistente / Infecciones recurrentes de piel, mucosas, tracto respiratorio o urinario / Úlceras,
gingivitis / Sepsis y cuadros graves sin signos inflamatorios evidentes
- Neutropenia congénita: Infancia, infecciones graves tempranas.
CLASIFICACIÓN
Duración y etiología
| Duración | Mecanismo |
| Aguda -> Días - semanas | Congénita / Adquirida |
| Crónica -> >3 meses | Benigna / Patológica |
NEUTROPENIA CICLICA
- Autosómica dominante / Mutación ELANE (19q13) / Episodios /21 días de neutropenia grave (3-6 días).
- Síntomas: Fiebre / Anorexia / Úlceras orales / Linfadenopatía cervical.
DIAGNÓSTICO
- Hemograma con fórmula leucocitaria
- Estudio de médula ósea (Aspirado y biopsia) -> Si hay pancitopenia o sospecha de displasia / Maduración detenida.
- Serología virales y pruebas autoinmunes.
- Vitaminas (B12 y folato).
- Pruebas genéticas (Casos congénitos o persistentes).
- Estudio de anticuerpos antineutrófilos -> Sospecha de neutropenia inmune.
HISTOLOGÍA
Médula ósea
- Hipoplasia granulocítica si hay defectos en producción.
- Maduración detenida (Como Kostmann).
- Puede haber displasia -> Sx mielodisplásicos.
- Infiltración medular -> Neoplasias secundarias.
TRATAMIENTO
- Tratar la causa subyacente -> Suspender fármaco -> Tratar infección -> Corregir déficit.
- Antibióticos de amplio espectro -> Neutropenia febril.
- Factores de crecimiento hematopoyético (G-CSF/filgrastim) -> Neutropenia congénita severa / Neutropenia febril postquimioterapia.
- Inmunoglobulina IV -> Casos autoinmunes.
- Trasplante de médula ósea -> Curativa -> Neutropenias congénitas graves (que no responden a G-CSF.
- Seguimiento continuo -> Crónicos.
TROMBOCITOPENIA
- Plaquetas en sangre periférica <100,000.
- <50,000 plaquetas/mm3: Riesgo de sangrado significativo.
- <20,000 plaquetas/mm3: Sangrado espontáneo.
Dx de confirmación -> Frotis de sangre periférica -> Descartar Pseudotrombocitopenia por microagregados plaquetarios.
ETIOPATOGENIA
- Disminución de la producción plaquetaria (origen central).
- Disminución del número de megacariocitos: Infiltración de médula ósea (leucemias, linfomas) / Aplasia medular / Sx de Fanconi / Trombocitopenia cíclica / Infección por rubéola congénita.
- Trombopoyesis ineficaz: Sx de Wiskott-Aldrich / Anemias megaloblásticas (déficit de B12 o ácido fólico).
- Aumento en la destrucción plaquetaria (origen periférico).
- Inmunitaria: PTI / LES / Linfomas / VIH.
- No inmunológicas: Fármacos / Púrpura post transfusional.
- Secundaria a hiperesplenismo
- Aumento compensatorio de megacariocitos en médula ósea.
PÚRPURA TROMBOCITOPÉNICA INMUNITARIA (PTI)
- Forma aguda -> Niños / Inicio súbito posterior a infecciones virales respiratorias / Resolución espontánea en semanas / Se asocia a eosinofilia y linfocitosis.
- Forma crónica (Enf de Werlhof) -> Adultos jóvenes (Mujeres)
PATOGENIA
- Presencia de autoanticuerpos lgG dirigidos contra glucoproteínas plaquetarias (Ib, Ilb/Illa)
- Unión del anticuerpo -> Opsonización -> Destrucción en el bazo por macrófagos.
- Activación del sistema del complemento.
DIAGNÓSTICO
- Confirmación de trombocitopenia -> Por descarte
- Frotis de sangre periférica.
- Estudio de médula ósea -> Sospecha de hipoproducción.
TRATAMIENTO
Se basa en el grado de trombocitopenia y presencia de sangrado:
- < 20,000 plaquetas/mm3 o con sangrado activo -> Tto inmediato.
- 20,000 - 50,000 plaquetas/mm3: Riesgo de sangrado o procedimientos invasivos.
- >30,000 plaquetas/mm3 sin factores de riesgo -> Observación.
1º línea -> Corticoides / Inmunoglobulina IV (grave o urgencia).
2º línea -> Esplenectomía / Rituximab / Inmunosupresores: Azatioprina, ciclofosfamida, micofenolato, ciclosporina A / Agentes citotóxicos: Vincristina.
LEUCEMIA LINFOBLÁSTICA AGUDA
Neoplasia Linfoproliferativa, proliferación descontrolada, potencial replicativo, bloqueo de la apoptosis.
- 4-5 años (+ común), 50 años.
- Tipo T -> Más malas
- Tejido linfoide primario: Timo y médula ósea.
- Tejido linfoide secundario: Bazo, ganglios.
Sustituye todo por células inmaduras.
- Leucemia de cél. Pequeñas (núcleo pequeño)
L1 + común.
- Poco citoplasma; Escasos núcleos.
- Leucemia de células grandes (núcleo grande)
>
L2
- + nucleolos; Irregulares.
- Leucemias vacuoladas
- Tipo burkitt.
→ L3 + grave.
Entre + inmaduro sea -> Van a expresar marcadores comunes. Entre + maduro -> + antígenos superficie
Síntomas generales
- Sx anémico (normocítica, normocrómica).
- Sx purpúrico (plaquetas bajas).
- Sx infiltrativo y metabólico (por infiltración de la médula ósea)
- Dolor óseo, especialmente en el esternón y huesos largos.
- Dolor articular.
- Linfadenopatía (ganglios linfáticos aumentados, no dolorosos).
- Hepatoesplenomegalia.
- Leucopenia o leucocitosis.
- Aumento de linfocitos y monocitos.
- Neutropenia severa (<500 células/uL): Riesgo de infecciones graves.
LCR -> Líquido turbio; Si hay >20% de blastos, se confirma leucemia aguda.
Factores de riesgo
- Estabilidad cromosómica (1-9).
- Sexo femenino -> Mejor pronostico.
Linfocitosis
Clasificación OMS
- Pronóstico: Importante considerar.
- Cromosoma
Filadelfia:
Impacto en el
pronóstico.
- <50,000 linfocitos/uL:
- 100,000 linfocitos/uL: Peor pronóstico, especialmente con infiltración al SNC.
- VCR: Aumento en la proliferación celular.
- ABL: Inhibe la apoptosis (+ resistencia al tto).
Diagnóstico
- Aspirado de médula ósea: fundamental para confirmar el diagnóstico.
Tratamiento indicaciones
- Inducción: 4-6 semanas para alcanzar remisión.
¿Non has encontrado lo que buscabas?
Explora otros temas en la Algor library o crea directamente tus materiales con la IA.
