Farmacodinamica: curve concentrazione-risposta e antagonisti
Documento della Prof.ssa Chiara Ruzza su farmacodinamica. Il Pdf esplora la relazione tra curva concentrazione-risposta e curva di binding, il comportamento degli antagonisti competitivi e non competitivi, inclusi gli agonisti parziali, per lo studio universitario della farmacologia.
Mostra di più16 pagine


Visualizza gratis il Pdf completo
Registrati per accedere all’intero documento e trasformarlo con l’AI.
Anteprima
Farmacodinamica: Curva Concentrazione-Risposta e Curva di Binding
Farmacologia, Lezione 03, 07/03/2023
Prof.ssa Chiara Ruzza
FARMACODINAMICA
Relazione tra curva concentrazione-risposta e curva di binding
La professoressa afferma che ripartirà dall'argomento trattato nella lezione precedente riguardante la relazione tra la
curva concentrazione e risposta e la curva di binding.
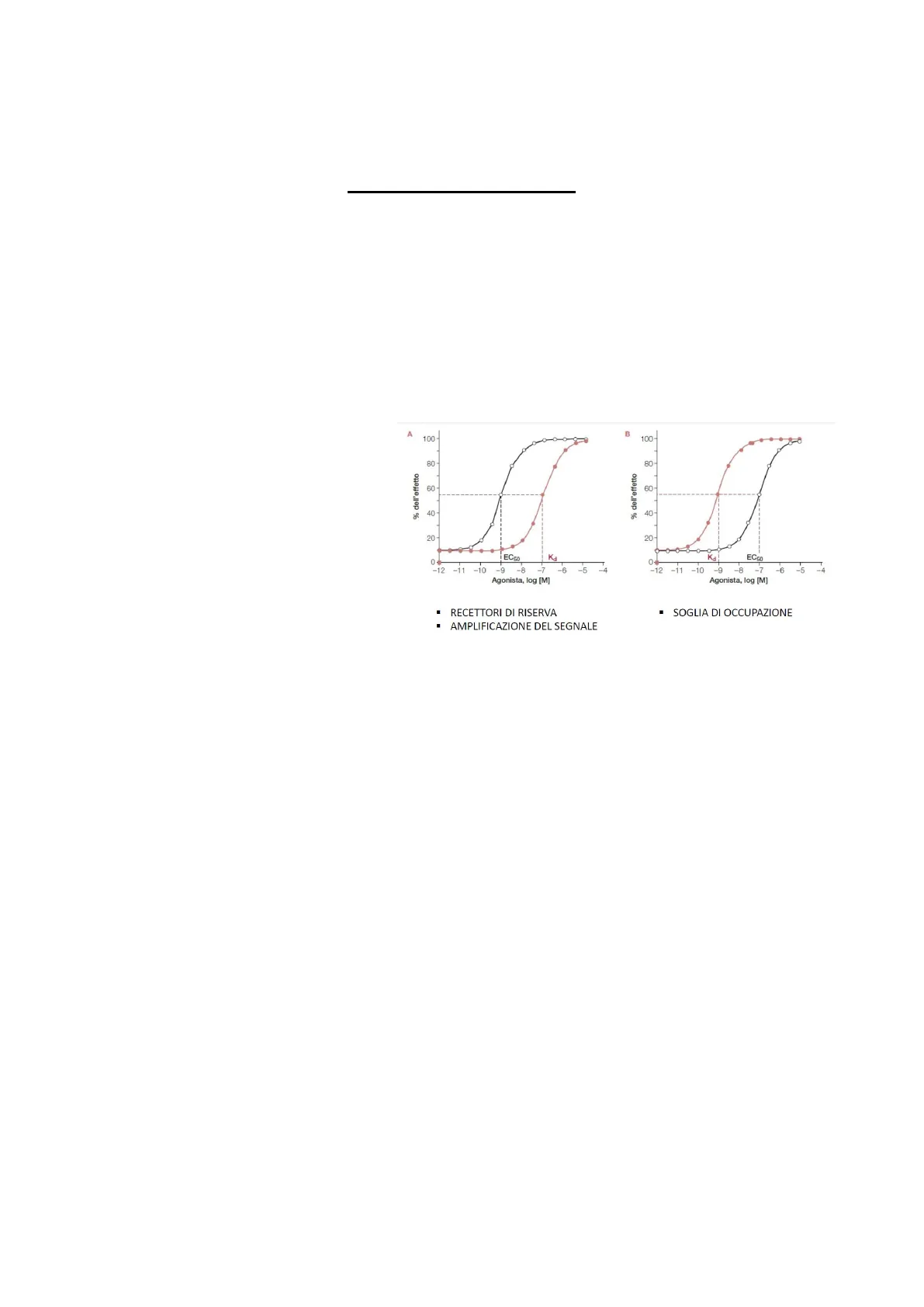
Un tempo la teoria dell'occupazione recettoriale sosteneva che la curva concentrazione-risposta registrata in
un saggio funzionale per un agonista fosse sovrapposta alla curva di binding, ossia che, nel momento in cui
vengono occupati tutti i recettori a disposizione, si ottiene l'effetto massimo e la Kd coincide con la EC50.
Adesso sappiamo che non è così, dato che
A
100
B
100-
sono stati fatti numerosi esperimenti di
80
80
binding che sono stati confrontati con
60
60
diversi esperimenti di agonismo in saggi
40
40
% dell'effetto
% dell'effetto
funzionali. Sappiamo che a volte le due
20
20
K
0
-7
ECCO
Ka
EC50
-5 -4
curve sono sovrapposte, a volte la curva
-12 -11 -10
-9
-8
-6
0
-12 -11 -10 -9 -8 -7 -6 -5 -4
Agonista, log [M]
Agonista, log [M]
concentrazione-risposta in un saggio
RECETTORI DI RISERVA
· SOGLIA DI OCCUPAZIONE
funzionale si trova a sinistra della curva di
AMPLIFICAZIONE DEL SEGNALE
binding (quest'ultima è rosa nell'immagine) mentre a volte succede il contrario.
Nell'immagine a destra si vede infatti in rosa la curva di binding posizionata a sinistra della curva
concentrazione-risposta in saggio funzionale. Quindi, la teoria dell'occupazione recettoriale non è del tutto
corretta ma va modificata.
Curva Concentrazione-Risposta a Sinistra della Curva di Binding
Per comprendere a cosa è dovuto questo comportamento analizziamo il grafico di sinistra, che rappresenta la
situazione più comune, ossia la curva concentrazione-risposta a sinistra della curva di binding. Un
comportamento di questo tipo ci indica che basta aver occupato (prendendo il caso in esempio) circa il 20%
dei recettori per ottenere una risposta quasi massimale. Basta occupare circa il 10% dei recettori per essere già
all'EC50. Questo può avvenire per due fenomeni principali, tra cui la presenza di recettori di riserva, ossia ci
sono più recettori di quelli necessari per generare la massima risposta che quella cellula o tessuto può produrre.
Questo significa che è sufficiente occupare un numero più basso di recettori per raggiungere l'effetto massimo
producibile dal sistema. Quindi, le cellule spesso presentano recettori di riserva, è una cosa assai comune e
dipende dal sistema che sto andando a studiare. L'altro fenomeno per cui la curva concentrazione-risposta è a
sinistra della curva di binding è l'amplificazione del segnale. Se consideriamo per esempio l'attivazione di
un recettore accoppiato a proteine G, a partire dall'attivazione del recettore da parte dell'agonista e andando
verso l'interno della cellula nei vari step della cascata del segnale, avremo un segnale che viene sempre più
1amplificato. Più ci mettiamo nelle condizioni sperimentali di studiare un segnale amplificato e più vedremo
questa curva concentrazione-risposta a sinistra della curva di binding.
Esempio di Amplificazione del Segnale: Via del Glucagone
Per fare un esempio di ciò che è appena stato riportato guardiamo la via di
signaling del glucagone che porta alla glicogenolisi, quindi alla produzione
di glucosio a partire dal glicogeno. Il glucagone (molecola agonista) attiva
un recettore, viene reclutata e attivata una proteina G, che a sua volta attiva
diverse adenilato ciclasi. Perciò, scendendo nella cascata del segnale,
abbiamo un primo step di amplificazione. Una singola adenilato ciclasi
produce tante molecole di AMP ciclico e ognuna di queste va ad attivare una
proteina chinasi A, quindi avremo molte molecole di AMP ciclico che
indicano l'attivazione di tante proteine chinasi A. A loro volta, le PKA attive
saranno in grado di fosforilare un numero maggiore di altre proteine chinasi.
Via via che scendiamo nella cascata del segnale e arriviamo alla fine della
via di signaling abbiamo un segnale che viene amplificato a cascata in
maniera esponenziale.
Glucagone
Adenilato
ciclasi
INATTIVA
Adenilato
ciclasi
ATTIVA
CAMP
ATP
Proteinchinasi
INATTIVA
Proteinchinasi
ATTIVA
ADP4
P
ATP
Fosforilasi
chinasi
ATTIVA
P
Fosforilasi
chinasi
INATTIVA
ATP
+ADP
Fosforilasi
INATTIVA
Fosforilasi
ATTIVA
P
Glucosio
Glicogeno
Conseguenze dell'Amplificazione del Segnale nello Studio degli Agonisti
La diretta conseguenza di ciò, nel momento in cui si vuole studiare l'effetto di un agonista, dipende dal tipo di
evento che si va ad analizzare. Se si sceglie un saggio funzionale in cui si studia un evento che è molto vicino
all'attivazione del recettore, ad esempio il GTPyS, l'amplificazione è pressoché nulla e quindi si avranno meno
artefatti dovuti ai fenomeni di amplificazione del segnale. Di conseguenza, la curva concentrazione-risposta
sarà molto simile alla curva di binding. Se, invece, si sceglie di misurare come evento intracellulare il livello
di AMP ciclico, si avrà una certa amplificazione del segnale e quindi si vedrà la curva concentrazione-risposta
spostata a sinistra rispetto alla curva di binding. Se si decide infine di misurare i livelli di produzione del
glucosio, quindi l'ultimo step di questa via del signaling, in cui si ha la massima amplificazione del segnale,
quello che si vedrà sarà una curva concentrazione-risposta spostata decisamente a sinistra rispetto alla curva
di binding.
Dunque, le due motivazioni di questo fenomeno sono i recettori di riserva oppure un saggio funzionale che
prevede una certa amplificazione del segnale. È molto comune questo perché la misura dei livelli di AMP
ciclico o la misura dei livelli di calcio intracellulare vengono utilizzati tantissimo per studiare ad esempio le
vie di signaling dei recettori accoppiati a proteine G. Quando si studia la farmacologia di un composto bisogna
conoscere il sistema biologico, quindi sapere se si sta lavorando con cellule che esprimono tanti recettori e
questi ultimi potrebbero anche essere sovra espressi. Allo stesso tempo, bisogna conoscere le caratteristiche
del test del saggio che si sta utilizzando. Nel caso di un GTPyS non si ha amplificazione, mentre con altri saggi
si possono avere diversi livelli di amplificazione. In definitiva, bisogna conoscere molto bene il substrato
biologico, il test e la misura biologica che si andrà ad analizzare.
2
Curva Concentrazione-Risposta a Destra della Curva di Binding
A destra si vede la situazione opposta, che è quella che avviene di rado e di cui non si conoscono bene le
ragioni biologiche. In questo caso, con un binding del 20% dei recettori disponibili, non si vede ancora alcuna
risposta biologica, che si può invece cominciare a vedere dopo avere occupato già l'80/90% dei recettori.
Significa che questo sistema richiede una soglia di occupazione recettoriale prima di poter produrre un effetto
biologico, cioè i primi recettori vengono occupati ma ciò non è sufficiente a generare una risposta.
Con queste ultime due slide l'insegnante afferma di aver concluso la parte della metodica di studio di un farmaco agonista.
Ora inizierà la trattazione dei farmaci antagonisti.
Farmaci Antagonisti
I farmaci antagonisti sono farmaci che legano il recettore ma non producono alcun effetto biologico, quindi
sono farmaci che possono essere studiati al binding perché hanno affinità per un certo recettore, di cui si può
calcolare la Kd (costante di dissociazione). Se però si decide di studiarli in un saggio funzionale, facendo una
concentrazione-risposta, si vedrà una riga piatta e il motivo è che questi farmaci legano il recettore e ne
stabilizzano la conformazione inattiva, non sono quindi dotati di efficacia.
Gli antagonisti possono essere dei farmaci, quindi considerati utili dal punto di vista clinico, perché occupano
il sito di legame del recettore impedendo il legame con l'agonista endogeno. In farmacologia speciale si
vedranno molti esempi, tra cui gli antagonisti dei recettori beta-adrenergici, alfa-adrenergici, antagonisti del
recettore dell'angiotensina 1.
Gli antagonisti possono interagire con il sito di legame per l'agonista in due modi, in maniera competitiva o
sormontabile (quest'ultimo termine lo si può trovare nei testi di farmacologia) oppure in maniera non
competitiva o insormontabile.
Antagonista Competitivo (Sormontabile)
Un antagonista competitivo lega esattamente lo stesso sito di
legame dell'agonista endogeno, ad esempio un beta-bloccante
competitivo lega il recettore beta nello stesso sito in cui lo
lega la noradrenalina. Il legame che forma l'antagonista
competitivo con il sito di legame del recettore è un legame di
tipo reversibile. Significa che l'antagonista può legare il
recettore ma si può anche staccare e che le molecole
antagonista e agonista endogeno possono competere per
questo sito di legame.
ANTAGONISTA COMPETITIVO
(SORMONTABILE)
A
B
recettore
L'antagonista lega lo stesso sito recettoriale dell'agonista.
Quando due molecole si presentano allo stesso sito recettoriale
solo una delle due molecole può legarsi. Se il legame è
reversibile si instaurerà una «gara» (COMPETIZIONE) tra i due
ligandi per occupare il recettore. Il recettore sarà maggiormente
occupato dal ligando con maggiore affinità. Se a contrastare il
legame del farmaco affine mettiamo una grossa quantità del
farmaco poco affine aumentiamo la probabilità che il farmaco
poco affine leghi il recettore.
Se con A intendiamo l'agonista endogeno e con B il farmaco antagonista competitivo, nel sito di legame che
c'è sul recettore si instaura proprio una competizione. A parità di concentrazione, quindi con 1 nM di agonista
A e 1 nM di antagonista B, si legherà il composto che ha una maggiore affinità per il recettore. Però, poiché le
molecole competono, si può cambiare questa situazione lavorando sulla concentrazione. Se ad esempio ci si
3trova nella situazione per cui A è più affine al recettore rispetto a B, a parità di concentrazione, tende a legarsi
A, ma se si duplica, centuplica o si moltiplica per mille la concentrazione di B, a questo punto B sarà in eccesso
e sarà in grado di spostare A dal recettore e prendere il suo posto. Perciò, quando si parla di antagonismo di
tipo competitivo bisogna sempre immaginare che l'antagonista compete con l'agonista endogeno e bisogna
dunque immaginarsi un sistema all'equilibrio in cui la molecola di antagonista può legarsi e staccarsi così
come la molecola di agonista può legarsi e staccarsi. La molecola che lega il recettore sarà quella con maggiore
affinità ma si può sempre giocare sulla concentrazione per spostare questo equilibrio.
Antagonista Non Competitivo (Insormontabile)
Con un antagonista non competitivo, dal punto di vista
biologico e biochimico, possono succedere due cose
nell'interazione con il recettore. Uno dei due eventi che
A
può verficarsi è che l'antagonista leghi il recettore in
B
maniera irreversibile. Non si instaura una competizione
tra queste due molecole perché l'antagonista si è
L'antagonista lega un sito recettoriale diverso da quello
dell'agonista (SITO ALLOSTERICO) oppure l'antagonista lega lo
stesso sito recettoriale dell'agonista ma in maniera irreversibile. Il
recettore occupato da un farmaco irreversibile non è più
disponibile. In questi casi non può esserci competizione tra le due
molecole per il legame con il recettore.
attaccato, non si stacca più e quel recettore non sarà mai
più disponibile per il legame con l'agonista endogeno.
Affinché l'agonista endogeno possa trovare altri recettori
occorre che la cellula ne sintetizzi di nuovi. L'altra cosa che può succedere è che l'antagonista sia non
competitivo perché lega un sito di legame diverso da quello dell'agonista endogeno (questa situazione non è
rappresentata nell'immagine). Dunque, non si ha competizione tra le due molecole per lo stesso sito recettoriale
semplicemente perché l'antagonista si è recato da un'altra parte e quindi non può essere spiazzato dall'agonista.
Questo comportamento viene anche chiamato modulazione allosterica negativa perché in questo caso
l'antagonista lega un sito allosterico. Abbiamo già utilizzato questo termine per parlare dei recettori canale. Il
sito ortosterico è quello in cui si lega il ligando endogeno, mentre il sito allosterico (o siti allosterici, dato che
ce ne possono essere più di uno) sono dei siti di legame diverso.
ANTAGONISTA NON COMPETITIVO
(INSORMONTABILE)
Riassunto Antagonismo Competitivo e Non Competitivo
Riassumendo:
Per quanto riguarda l'antagonismo competitivo, l'antagonista lega lo stesso sito di legame dell'agonista
endogeno in maniera reversibile. Si instaura la competizione tra le due molecole per il sito di legame.
Nell'antagonismo non competitivo i casi sono due. Si formano dei legami di tipo irreversibile tra l'antagonista
e il recettore e quindi questo antagonista non sarà mai più disponibile per l'agonista endogeno oppure ci può
essere un sito allosterico di legame e in questo caso possiamo parlare anche di modulazione allosterica negativa.
È stato detto che l'antagonista non è dotato di efficacia, quindi ora è necessario capire come fare per studiarlo
nei saggi funzionali. L'unica possibilità è di studiarlo in presenza dell'agonista di riferimento, che
generalmente è l'agonista endogeno per il recettore, ma può essere anche un agonista noto o un agonista
standard per quel recettore. I protocolli di studio per i farmaci antagonisti competitivi e non competitivi sono
sostanzialmente gli stessi ma i risultati che si ottengono sono diversi.
4
Non hai trovato quello che cercavi?
Esplora altri argomenti nella Algor library o crea direttamente i tuoi materiali con l’AI.
