Fisiopatologia dell'insufficienza cardiaca: alterazioni cellulari e meccanismi
Documento universitario sulla fisiopatologia dell'insufficienza cardiaca. Il Pdf esplora le alterazioni cellulari, la morte cellulare, la fibrosi e i meccanismi neuro-ormonali, utile per lo studio della biologia a livello universitario.
Mostra di più12 pagine


Visualizza gratis il Pdf completo
Registrati per accedere all’intero documento e trasformarlo con l’AI.
Anteprima
Fisiopatologia dell'insufficienza cardiaca
Il professore inizia ripetendo gli ultimi argomenti della lezione precedente. Elenca le alterazioni biochimiche responsabili della crescente incapacità del cuore di rispondere ad un aumento del carico di lavoro. Riprende poi la spiegazione dal punto in cui si era interrotto la scorsa volta.
Alterazioni cellulari
Alterazioni cellulari.
- Espressione genica: per adattarsi alle condizioni di sovraccarico, il cuore inizia anche ad esprimere una serie di geni responsabili della proliferazione (c-myc, c-jun, c-fos). In questo caso, trattandosi di cellule permanenti, il risultato è l'aumento del numero dei sarcomeri (ipertrofia), le cui fibrille possono essere disposte sia in serie che in parallelo, in particolare isoforme fetali di miosina e troponina, con una scarsa capacità contrattile.
- morte cellulare
- fibrosi
- >> autofagia, mitofagia
Alterazioni cellulari
- omeostasi del calcio (<< RyR, << IP3R, << SR CaATPase)
- desensibilizzazione adrenergica (< espressione ß recettori, disaccoppiamento, espressione proteine G inibitorie)
- ipertrofia (>> c-fos, c-jun, c-myc, > numero dei sarcomeri, > miosina e troponina fetale)
- >> espressione di proteine fetali non contrattili
- << rapporto capillari/tessuto contrattile
Fattori causali e facilitanti l'ipertrofia patologica
Fattori causali e facilitanti l' ipertrofia patologica: > Citochine infiammatorie, angiotensina, stress emodinamico
- Morte cellulare: lo stress meccanico e metabolico a cui sono sottoposti i miocardiociti aumenta i fenomeni di apoptosi.
- Fibrosi: dovuta principalmente al rilascio extracellulare di TGF-beta, la secrezione di collagene diminuisce ulteriormente la contrattilità.
- Rapporto capillari/tessuto contrattile: questo aumento di volume dei miocardiociti non è seguito da un aumento del numero di capillari (riduzione rapporto), la scarsa ossigenazione del tessuto cardiaco impedisce una adeguata sintesi di ATP.
- Metabolismo energetico: il deficit energetico viene compensato con la degradazione dei componenti cellulari tramite l'autofagia, col procedere della malattia vengono anche degradati organelli come i mitocondri (mitofagia).
Fattori facilitanti l'ipertrofia patologica
1Fattori facilitanti l'ipertrofia patologica. Oltre al sovraccarico meccanico, intervengono altri fattori facilitanti l'ipertrofia patologica:
- L'infiammazione è uno di questi, il danno tissutale attiva l'infiammazione e quindi il rilascio di citochine infiammatorie della fase acuta (TNF-alfa, IL-1 e IL-6), queste ultime hanno un effetto cardiodepressivo. (il prof anticipa che quando verrà trattato lo shock, si studierà la SIRS "sindrome da risposta infiammatoria sistemica, e si vedrà come indipendentemente dal fatto che il danno interessi il tessuto cardiaco o meno, ci sarà sempre questo effetto cardiodepressivo delle citochine)
- Angiotensina: rilasciata a livello sistemico, agisce sul circolo arterioso e su quello capillare cardiaco, riducendo la perfusione coronarica.
- Stress emodinamico: un esempio è proprio l'ipertensione arteriosa, che è sia un fattore iniziale sia un fattore che permette l'aggravamento dell'ipertrofia andando a creare un circolo vizioso.
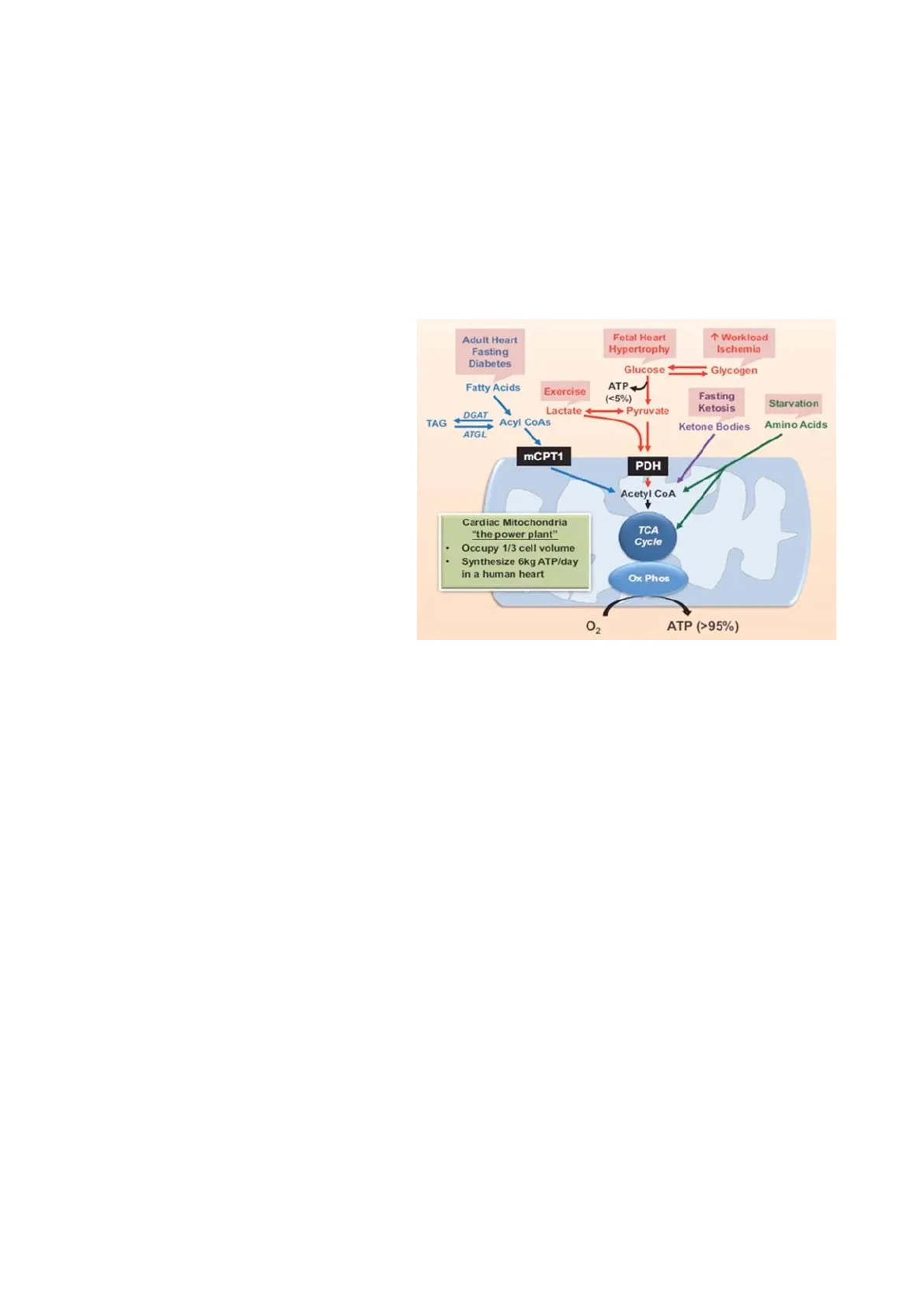
Adult Heart Fasting Diabetes Fetal Heart Hypertrophy + Workload Ischemia Glucose Glycogen Fatty Acids Exercise ATP (<5%) Pyruvate Fasting Ketosis Starvation DGAT Lactate TAG Acyl CoAs Ketone Bodies Amino Acids ATGL mCPT1 PDH Acetyl COA + Cardiac Mitochondria "the power plant" TCA Cycle · Occupy 1/3 cell volume · Synthesize 6kg ATP/day in a human heart 0x Phos - Riattivazione geni fetali: questi ATP (>95%) O2 pattern genetici vengono riattivati poiché i normali meccanismi di adattamento (tra cui una già aumentata sintesi di proteine dell'adulto) non sono sufficienti a compensare l'aumento di lavoro permanente. Questi prodotti, come troponina e miosina fetale, non sono però compatibili col cuore adulto, in quanto il loro scopo era la replicazione dei miocardiociti. Questo aggrava ulteriormente il quadro già compromesso, poiché la risposta di adattamento indotta non è efficace. - Riprogrammazione metabolica: in condizioni normali il cuore soddisfa il 60% del suo fabbisogno energetico con la beta-ossidazione degli acidi grassi. Un cuore ipertrofico necessita di un quantitativo di energia e ossigeno maggiore a causa dell'aumento della massa, in più non vi è un aumento adeguato di capillari (il cuore ipertrofico diventa anche vulnerabile allo scompenso associato all'ischemia, che può condurre alla morte. Robbins 2 pag 510 IX ediz.). Inizialmente, per far fronte a questa maggior richiesta, il glucosio e il lattato divengono i principali substrati del metabolismo energetico. Nel prosieguo dello stato di sofferenza in corso di ipertrofia patologica, questi substrati diventano insufficienti, quindi entrano in atto fenomeni autofagici e mitofagici, per supplire momentaneamente al deficit energetico.
Autofagia
2Autofagia. In questo caso si attiva un circolo vizioso che si auto alimenta e aggrava le condizioni di deficit energetico. Le condizioni di scarsa ossigenazione e la mancanza di substrati energetici inducono stress mitocondriale, quindi diminuisce ancora di più la produzione di ATP e aumenta quella di ROS. La diminuzione di ATP, contemporaneamente all'aumento di AMP, attiva la chinasi AMPK che è uno dei principali attivatori dell'autofagia. Anche i ROS attivano l'autofagia, oltre a danneggiare componenti cellulari. Viene letta l'immagine ripetendo alcuni concetti già spiegati, aggiungendo che, nello stadio di ipertrofia patologica, se i meccanismi di compenso funzionano non si instaura lo stadio di scompenso cardiaco congestizio (spiegato dopo). Lo scompenso si manifesterà se non vengono risolte le cause che hanno portato all'ipertrofia.
Autofagia e insufficienza cardiaca
Autofagia ed insufficienza cardiaca AMPK Į ATP 1 autophagy ÎROS protein damage / misfolding energy crisis mitochondrial tarnotina Patologia Il e fisiopatologia - Lezione 22 Fig. 4. Metabolic functions of autophagy in heart failure. During the progression from adaptive cardiac hypertrophy to decompensated heart failure, autophagy can be induced by accumulation of reactive oxygen species (ROS) and by oxidized and misfolded proteins. Enhancement of autophagic activity, in turn, can target mitochondria selectively (mitophagy) or indiscriminately. thereby negatively impacting ATP production. exacerbating ROS generation, and promoting disease progression I livelli di autofagia possono aumentare fino a digerire completamente interi organelli, soprattutto i mitocondri (mitofagia). Si instaura così una crisi energetica.
Autofagia, ipertrofia e scompenso cardiaco
Autofagia ed insufficienza cardiaca La progressione verso lo scompenso Cuore normotrofico o in corso di ipertrofia fisiologica Cuore in corso di ipertrofia patologica Cuore in corso di scompenso phenotype metabolic profile fatty acids glucose fatty acids glucose fatty acids glucose Patologia Il e fisiopatologia - Lezione 22 autophagy homeostasis 1 provision mitochondrial targeting? outcome Fig. 5. Metabolic profit and loss of autophagy in cardiac hypertrophy and failure. Basal autophagic activity is essential to maintain cardiomyocyte energy production and cellular homeostasis. The significant increase in autophagy seen during cardiac hypertrophy may contribute to adaptive metabolic remodeling which together with enhancement of glucose utilization plays critical roles in ATP production and macromolecule biosynthesis required for hypertrophic growth. The deterioration of energy status in end-stage heart failure triggers exuberant autophagy which may target mitochondria for degradation finther impairing hoth glucose and fatty acid metabolism
Insufficienze cardiache
3Insufficienze cardiache. L'insufficienza cardiaca è ventricolare, scolasticamente Si parla di insufficienza destra e sinistra anche se entrambe convergono nell'insufficienza d'organo. Entrambe possono essere dovute sia da deficit diastolico, ovverosia ridotto riempimento ventricolare, sia deficit sistolico, ovvero ridotta efficienza della contrazione ventricolare. Anche in questo caso entrambe convergono nel quadro generale di insufficienza ventricolare. Le cause possono essere molteplici, di cui le più importanti sono sovraccarico emodinamico e sovraccarico pressorio.
Insufficienza ventricolare sinistra
Insufficienza ventricolare sinistra Deficit sistolico da ridotta efficienza di contrazione ventricolare Ma in genere sono associati Cause: Sovraccarico emodinamico (insufficienza mitrale o aortica, insufficienza ad alto volume d' elezione da anemie o ipertiroidismo). Sovraccarico pressorio (ipertensione sistemica, ostruzioni valvolari, per es, aortica). Danno miocardico con perdita di tessuto (infarto, connettivopatie, per es. LES). Danno miocardico da tossici o infezioni (etilismo, doxorubicina, virus, batteri) Ridotto riempimento (stenosi mitralica, pericarditi o tamponamento cardiaco, malattie infiltrative, per es. amiloidosi, disturbi del ritmo, per es. fibrillazione atriale).
- Il sovraccarico emodinamico è dovuto a insufficienza valvolare e insufficienza ad alto volume (in particolare della sinistra, ma anche della destra indirettamente). Quest'ultima è importante da ricordare; normalmente associamo l'insufficienza ventricolare ad una riduzione del volume sistole (GS), ma in caso di anemie o ipertiroidismo possiamo avere insufficienza anche con una gittata sistolica maggiore del normale. La definizione di insufficienza cardiaca è l'incapacità del cuore di sopperire alle esigenze metaboliche dei tessuti periferici. Considerando le due condizioni sopracitate: nell'anemia abbiamo un deficit di trasporto di ossigeno da parte del sangue (ipossia tessuti periferici) e allo stesso tempo una diminuita viscosità (bassa concentrazione di globuli rossi) che causa aumento del precarico e quindi della gittata, nell'ipertiroidismo abbiamo un aumentato metabolismo basale e si ha maggiore richiesta di O2. Quindi nonostante la gittata aumenti, non vengono soddisfatte le esigenze metaboliche dei tessuti.
- Il sovraccarico pressorio è dovuto a ipertensione arteriosa sistemica e ostruzioni valvolari (stenosi aortica).
- Lesioni del miocardio con perdita di tessuto come eventi infartuali o le connettivopatie, tra le quali rientra il lupus eritematoso sistemico (LES), o tutte le condizioni che portano alla sostituzione del tessuto cardiaco con un altro tessuto di origine istologica differente privo di attività contrattile.
- Danno del miocardico da tossine, infezioni (miocarditi), ed anche farmaci, ad esempio la doxorubicina, farmaco antiblastico usato nella terapia di svariate forme tumorali, molto efficiente ma gravato da un'importante cardio tossicità.
- Deficit diastolico, quindi uno scarso riempimento, può essere dovuto a stenosi mitralica e pericarditi, in quest'ultimo caso si verifica il fenomeno del tamponamento cardiaco, che causa difficoltà nell'espansione del ventricolo sinistro per aumento di pressione nella cavità pericardica
Deficit diastolico e cuore insufficiente
4 Deficit diastolico da ridotto riempimento ventricolare Cuore insufficiente
Non hai trovato quello che cercavi?
Esplora altri argomenti nella Algor library o crea direttamente i tuoi materiali con l’AI.
