Oncologia Medica: amiloidosi AL e sindromi mielodisplastiche
Documento di Oncologia Medica sull'amiloidosi AL e le sindromi mielodisplastiche. Il Pdf esplora l'amiloidosi AL, una discrasia plasmacellulare, e le sindromi mielodisplastiche, patologie del midollo osseo che causano citopenie, con classificazioni basate sul tipo di citopenia e numero di blasti.
Mostra di più13 pagine


Visualizza gratis il Pdf completo
Registrati per accedere all’intero documento e trasformarlo con l’AI.
Anteprima
ONCOLOGIA MEDICA
Prof. Caracciolo Lezione 36 - 02/05/2023 Sbobinatori: Irene Iannello (I ora), Martina Logiacco (II ora) Controllore: Alessia Morelli Argomenti: Amiloidosi, Sindromi Mielodisplastiche.
AMILOIDOSI
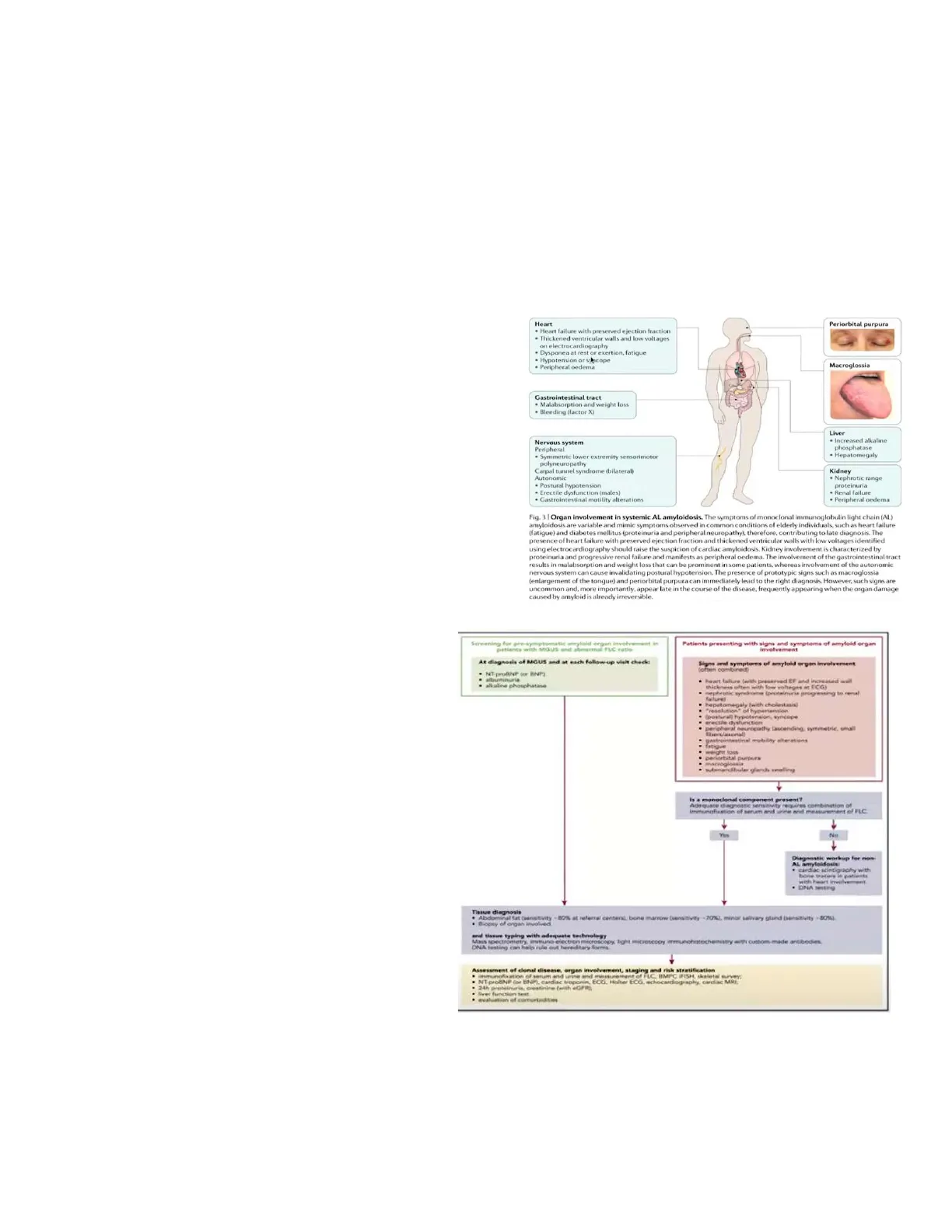
Oggi parliamo di un'altra discrasia plasmacellulare che è l'amiloidosi AL (light chain cioè catene libere). L'amiloidosi di per sé non è una discrasia plasmacellulare, ma è un gruppo di malattie complesse caratterizzate dal missfolding, quindi dall'alterato ripiegamento delle proteine e soprattutto della loro aggregazione in fibrille di amiloide che si depositano nei tessuti risultando in un progressivo danno d'organo. Nell'ambito della amiloidosi ci sono 36 diverse proteine che possono essere causa dell'amiloidosi. L'amiloidosi che interessa a noi è l'amiloidosi AL che è la prima mostrata nella slide (immunoglobulin light chain). Le principali sono le immunoglobulin light chain, quella dovuta ad una aumentata produzione di catene libere, dovuta a un clone di plasmacellule (è questa la amiloidosi da discrasia plasmacellulare). L'altra frequente è l'amiloidosi da transtiretina che può essere ereditaria o acquisita > questa è la forma di amiloidosi dell'anziano. Poi c'è l'amiloidosi AA dove A sta per proteina A (che è una proteina di fase acuta che si accumula come secondaria ai processi infiammatori cronici) e si riferisce all'amiloidosi che si associa alle malattie infiammatorie croniche. Poi ci sono tantissime altre forme diverse che si associano al tipo di proteina che è responsabile della formazione dell'amiloide. Come vedete gli organi coinvolti sono diversi ma soprattutto il cuore, il rene, fegato, il sistema nervoso periferico e il tratto gastrointestinale. Ci sono delle forme di amiloide che possono essere sistemiche ma anche localizzate; difatti la malattia di Parkinson può essere considerata una forma localizzata di amiloidosi a livello del cervello.
Meccanismo dell'amiloidosi AL
Quindi quello che succede nell'amiloidosi AL è che c'è un piccolo (perché l'infiltrato è sotto il 10%) clone di plasmacellule monoclonali che producono soprattutto queste catene leggere che sono instabili da un punto di vista termodinamico e che per via di interazioni cellulari ed extracellulari e anche per blocco di proteine di degradazione precipitano sottoforma di oligomeri e l'associazione ad alcune proteine come la proteina SAP che blocca la degradazione di queste fibrille si accumulano e si assemblano a formare le fibrille di amiloide che con la colorazione rosso congo hanno questa birifrangenza verde al microscopio ottico, che ci permette di fare diagnosi di amiloidosi e che per una azione di massa e per una tossicità danno una disfunzione d'organo. Gli organi più coinvolti sono cuore e rene e raramente il fegato, il sistema nervoso sistemico e autonomo e periferico.
Sintomi e segni dell'amiloidosi AL
Il cuore dà segno di sé: con insufficienza cardiaca, con frazione di eiezione conservata (questa è la principale differenza con le insufficienze cardiache da cardiopatia ischemica), l'insufficienza cardiaca è dovuta ad un alterato rilasciamento diastolico; con ispessimento del setto interventricolare, con bassi voltaggi all'elettrocardiogramma, con ipotensione, sincope, dispnea, fatigue. La macroglossia è tipica. Il tratto gastrointestinale presenta malassorbimento, perdita di peso, disturbi della motilità intestinale. Il sistema nervoso dà segni di polineuropatie, la sindrome del tunnel carpale. Il fegato presenta epatomegalia e colestasi, mentre il rene presenta sindrome nefrosica fino all'insufficienza renale.
Diagnosi di amiloidosi
Quando vi trovate di fronte a questo pz con queste caratteristiche, dovete sempre sospettare una amiloidosi. Da un punto di vista diagnostico quello che dobbiamo capire è escludere le principali due cause, ovvero, o amiloidosi AL associata ad una discrasia plasmacellulare o una amiloidosi da transtiretina. La prima cosa da chiederci se ci sono questi segni e sintomi di compromissione d'organo potenzialmente da amiloide e bisogna capire se è presente una componente monoclonale. Se è presente una componente monoclonale a quadro siero- proteico ci orientiamo verso una amiloidosi AL.
Passaggi diagnostici
I passaggi diagnostici consistono nel rilevare essenzialmente: l' amiloide nel tessuto coinvolto. Non sempre è facile fare una biopsia renale o cardiaca o epatica. Quello che si può usare è un surrogato. ossia l'aspirazione del grasso addominale. Si fa, quindi, una aspirazione con la siringa e si va alla ricerca di amiloide attraverso la colorazione di rosso congo. In caso di difficoltà ad eseguire una biopsia del grasso addominale, si può fare anche quella delle ghiandole salivari minori, che hanno una sensibilità
Non hai trovato quello che cercavi?
Esplora altri argomenti nella Algor library o crea direttamente i tuoi materiali con l’AI.
