Fibrosi Cistica: cause, sintomi, complicanze e terapie attuali
Slide di Università sulla Fibrosi Cistica. La Presentazione descrive la fibrosi cistica, una malattia genetica ereditaria, analizzando cause, sintomi e complicanze. Il documento, utile per lo studio della Biologia, esplora le mutazioni genetiche e le nuove prospettive di cura, inclusi i farmaci che agiscono sul canale CFTR.
Mostra di più14 pagine


Visualizza gratis il Pdf completo
Registrati per accedere all’intero documento e trasformarlo con l’AI.
Anteprima
FIBROSI CISTICA
LEZIONE 3, 11/03/2025 Il termine scientifico è mucoviscidosi, anche se nel gergo comune fibrosi cistica è più utilizzato. Entrambe i termini comunque descrivono le caratteristiche di questa patologia: la fibrosi cistica è caratterizzata da cisti fibrotiche che sono delle caratteristiche morfologiche che si vengono a creare nei tessuti degli organi bersaglio di questa patologia. Queste cisti di tessuto fibrose sono il risultato di un'esagerata mucoviscidosi ecco perché appunto il termine fibrosi cistica e il termine scientifico mucoviscidosi.
La fibrosi cistica è una malattia genetica, la più diffusa al mondo, che ha come caratteristiche mutazioni nel gene CFTR. Le mutazioni a carico di questo gene, che è un canale, portano ad una serie di modificazioni per qui si viene a creare uno stato di mucoviscidosi. Ne deriva una produzione ed accumulo di muco denso, viscoso ed estremamente difficile da veicolare nei dotti principali, in particolare nei dotti respiratori, il che crea una sorta di ostruzione a cui consegue un ispessimento delle pareti.
L'accumulo di questo muco denso difficile da veicolare fa sì che si creino delle nicchie, ambiente favorevole per lo sviluppo di infezioni. I pazienti affetti da fibrosi cistica devono sottoporsi a un utilizzo cronico di antibiotici e di cortisone proprio perché sono più predisposti a sviluppare infiammazione. Questa nel tempo poi può diventare cronica e può richiamare degli infiltrati infiammatori, in particolare neutrofili, provocando un danno tissutale. Esso innescherà nuovi meccanismi di riparazione che cercheranno di riparare laddove il tessuto risulta danneggiato, modificandolo con del tessuto di sostegno mesenchimale che nel tempo porterà alla formazione di cisti fibrotiche.
Trasmissione della Fibrosi Cistica
La fibrosi cistica è una malattia genetica a trasmissione autosomica recessiva, questo significa che entrambi gli alleli del gene coinvolti in questa patologia devono essere mutati per lo sviluppo della patologia. Essa è la malattia genica più comune tra gli individui soprattutto di razza bianca: gli indici di incidenza ci dicono che 1:2500/3000 nati vivi ha la fibrosi cistica, quindi è una malattia molto diffusa nonostante venga trasmessa per via autosomica recessiva.
Si stima che almeno il 2-4% degli individui di razza bianca sia portatore della malattia quindi una persona su 25 è portatore della malattia. Essere portatori significa che uno dei due alleli del gene che esprime il canale CFTR porta delle mutazioni, quindi se due persone portatori di mutazioni del gene CFTR si accoppiano hanno:
- 25% di probabilità di avere un figlio del tutto sano
- 50% di probabilità di avere un figlio portatore anch'esso di mutazioni del gene
- 25% di probabilità di avere un figlio affetto da fibrosi cistica
GENE MUTATO GENE NORMALE 25% MALATO 50% PORTATORE SAND 25% SANO
Evoluzione della Fibrosi Cistica
Negli anni '60/'70 era una malattia prevalentemente pediatrica. La fibrosi cistica è una patologia con delle complicanze molto severe e soprattutto è una malattia sistemica (coinvolge più apparati), che in maniera preponderante colpisce i polmoni. La maggior parte dei pazienti affetti da fibrosi cistica moriva quindi per insufficienza respiratoria, con una aspettativa di vita molto bassa.
Con l'avanzare delle cure, lo sviluppo di nuove terapie e l'aumentare della qualità media della vita, dello stile di vita e anche delle condizioni igienico sanitarie oggi possiamo definire la fibrosi cistica come una patologia adulta, cioè i pazienti affetti da fibrosi cistica sono persone che arrivano a una mediana circa di 53 anni e arrivano anche alla vecchiaia.
Ancora non c'è una terapia per questo tipo di patologia ma le cure che ci sono attualmente disponibili hanno creato comunque una aspettativa di vita migliore e una condizione clinica sicuramente migliore rispetto a quelli che erano gli anni 70 e gli anni 80.
Diffusione della Fibrosi Cistica
Perché questa patologia è così diffusa nonostante sia una malattia genetica autosomica recessiva quindi entrambi gli alleli devono essere mutati affinché si sviluppi la patologia? Perché ha comunque un'incidenza di 1: 25.
Quest'incidenza è così alta perché nel corso della storia avere un'eterozigosi per questo canale CFTR era vantaggio selettivo, soprattutto in caso di pandemie, ad esempio, di colera o date da enterotossine batteriche. Mutazioni a carico di questo gene portano a muco denso, viscoso, disidratato. Il soggetto portatore di mutazioni, invece, è un soggetto sano che non manifesta alcun sintomo della patologia, ma tende ad avere un muco un po' meno idratato rispetto a una persona senza mutazioni e questo durante pandemie come ad esempio il colera, che porta a forte disidratazione e che non curata ha determinato la morte di milioni di persone, ha rappresentato un vantaggio.
Quindi le persone portatrici di questa mutazione in eterozigosi per il canale CFTR, se colpite da malattie enteriche o se colpite dal colera, rispondevano molto meglio rispetto a una persona sana. Questo, nel corso dell'evoluzione, ha fatto sì che venisse trasmessa, perché di fatto costituisce un vantaggio selettivo e una ulteriore protezione alle infezioni batteriche.
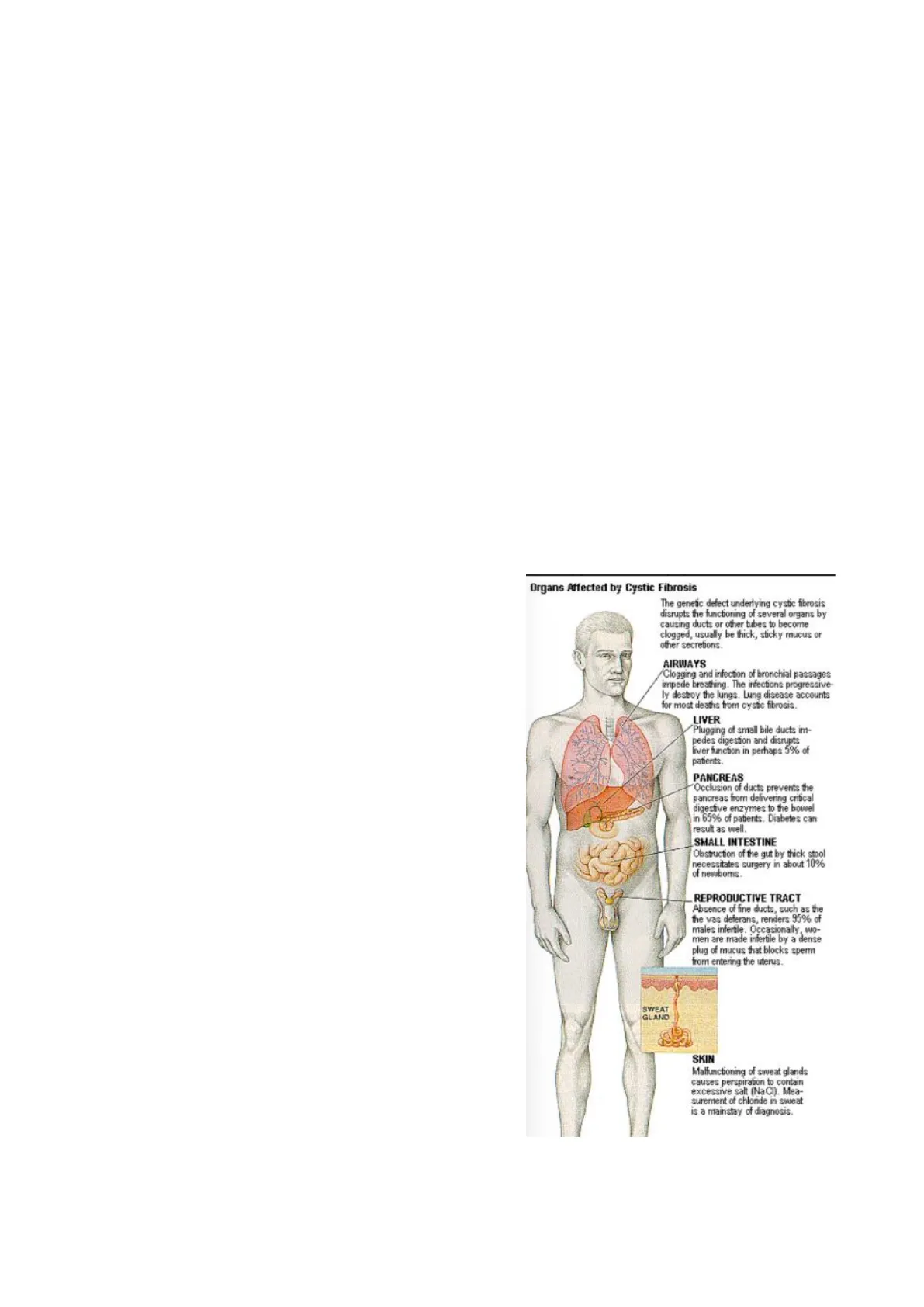
Organi Colpiti dalla Fibrosi Cistica
La fibrosi cistica è una malattia multisistemica che colpisce diversi organi, primo tra tutti il sistema polmonare: tutti i pazienti affetti da fibrosi cistica mostrano infezioni polmonari ricorrenti. È una malattia caratterizzata da alterazioni del processo secretorio di tutte le ghiandole esocrine, sia quelle muco-secernenti sia quelle sudoripare sparse a livello di tutto il corpo.
Si verifica la produzione di un muco viscido, denso, difficile da veicolare, disidratato che rimane all'interno dei dotti principali, non viene veicolato e provoca delle ostruzioni, responsabili dell'insorgenza delle manifestazioni cliniche tipiche delle malattie polmonari.
Un altro organo che è bersaglio di questo accumulo di queste secrezioni così dense e così disidratate è l'apparato gastrointestinale e possono essere presenti delle complicanze anche a livello della cute.
Il canale CFTR nelle ghiandole sudoripare lavora in modo contrario rispetto a tutte le altre cellule in cui è espresso.
Organs Affected by Cystic Fibrosis
The genetic defect underlying cystic fibrosis disrupts the functioning of several organs by causing ducts or other tubes to become clogged, usually be thick, sticky mucus or other secretions.
AIRWAYS
Clogging and infection of bronchial passages impede breathing. The infections progressive- ly destroy the lungs. Lung disease accounts for most deaths from cystic fibrosis.
LIVER
Plugging of small bile ducts im- pedes digestion and disrupts liver function in perhaps 5% of patients.
PANCREAS
Occlusion of ducts prevents the pancreas from delivering critical digestive enzymes to the bowel in 65% of patients. Diabetes can result as well.
SMALL INTESTINE
Obstruction of the gut by thick stool necessitates surgery in about 10% of newboms.
REPRODUCTIVE TRACT
Absence of fine ducts, such as the the vas deferans, renders 95% of males infertile. Occasionally, wo- men are made infertile by a dense plug of mucus that blocks sperm from entering the uterus.
SWEAT GLAND SKIN
Malfunctioning of sweat glands causes perspiration to contain excessive salt (NaCI). Mea- surement of chloride in sweat is a mainstay of diagnosis.
Infine tra gli organi interessanti ci sono l'apparato riproduttivo sia femminile che maschile. Essendo una malattia genetica colpisce indistintamente persone di sesso maschile o di sesso femminile. Essa si trasmette se due persone portatrici di mutazioni si accoppiano e hanno la probabilità del 25% di avere un figlio malato.
Test del Portatore per Fibrosi Cistica
Per sapere se si è portatore di mutazioni per il gene CFTR responsabile della patologia è possibile effettuare dei test del portatore. Questi vengono passati dal sistema sanitario nazionale e coprono una batteria dalle 34 alle 48 mutazioni che possono avvenire nel gene CFTR e che sono responsabili del circa 85% della fibrosi cistica.
CFTR: Regolatore della Conduttanza Transmembrana
Dal punto di vista eziologico la causa della fibrosi cistica sono mutazioni a livello del CFTR (cistis fibrosis transmembrane conductance regulator = regolatore della conduttanza transmembrana della fibrosi cistica). Questo gene si trova localizzato a livello del cromosoma 7, nel braccio lungo, e una volta trascritto dà origine a una proteina di circa 1500 aminoacidi.
La proteina, una volta che è stata tradotta, deve essere sottoposta ad una serie di modificazioni post traduzionali, necessarie per la sua corretta localizzazione e per la sua corretta funzione. In seguito matura, infine si ha un trafficking, viene veicolata a livello della superficie della membrana plasmatica delle cellule, dove viene espressa a livello apicale. Se vengono meno questi processi post traduzionali o se non si ha un corretto trafficking della proteina, essa non è in grado di svolgere la sua funzione e questo può portare alle conseguenze cliniche tipiche della fibrosi cistica, quindi tipiche della malattia.
Struttura del Canale CFTR
Come è formato questo canale? Principalmente è suddiviso in 5 domini:
2 domini transmembrana formati da 6 Alfa eliche, ciascuno che passa nella membrana plasmatica, sono responsabili della selettività del canale. Questi due domini transmembrana sono carichi positivamente, quindi il canale è anionico, selettivo per i bicarbonati e soprattutto per i cloruri, ioni carichi negativamente. I due domini transmembrana sono il core del canale perché ne permettono la selettività e si trovano localizzati a livello della membrana plasmatica.
MSD-1 MSD-2 Extracellular Plasma membrane Intracellular N- NBD-1 ATP NBD-2 ATP ADP + P ADP + P R domain C 1 PKA + ATP
Al di sotto di questi due domini transmembrana, a livello dello spazio citoplasmatico, troviamo 2 domini che legano nucleotidi, il che suggerisce che per funzionare correttamente il canale CFTR deve legare ATP. Essi infatti legano due molecole di ATP per far funzionare correttamente il canale.
Infine troviamo un dominio regolatorio che si trova sempre nello spazio citoplasmatico ed è responsabile anch'esso dell'apertura del canale. A livello di questo dominio regolatorio sono presenti dei siti di fosforilazione delle serine e delle treonine che sono bersaglio delle protein chinasi A. Quindi in seguito ad un aumento del secondo messaggero AMPciclico le proteine chinasi A vanno a bersagliare le serine e le treonine presenti sul dominio regolatorio e ne provocano l'apertura.
Riassumendo: affinché il canale si apra correttamente devono avvenire due eventi indispensabili: il legame con due molecole di ATP e la fosforilazione da parte della pKa. Se anche solo uno di due di questi eventi non avviene il canale non si apre correttamente. La pKa si attiva in seguito ad un aumento di AMPciclico come secondo messaggero e va a fosforilare delle serine e delle treonine che sono presenti a livello di questo dominio regolatorio. Una volta che quindi si ha la fosforilazione da parte della pKa e il legame con due molecole di ATP, si ha un cambiamento conformazionale, per cui il dominio regolatorio si allontana dai due domini transmembrana e permette il passaggio di ioni cloruro.
Il canale in tutte le cellule (ad eccezione delle ghiandole sudoripare) lavora per estrudere gli ioni cloruro dallo spazio citoplasmatico al lume della ghiandola.
Il canale CFTR è un canale dei cloruri, è canale anionico che si trova espresso sul lato apicale della membrana plasmatica da quasi tutte le cellule, anche le cellule del sistema del sistema immunitario. In tutte le cellule
Non hai trovato quello che cercavi?
Esplora altri argomenti nella Algor library o crea direttamente i tuoi materiali con l’AI.
