Tecnología del DNA Recombinante "in Vitro" y Fermentación Industrial
Documento de Universidad sobre Tecnología del DNA Recombinante "in Vitro". El Pdf, un documento de Biología para Universidad, explora métodos de manipulación del DNA, incluyendo obtención, amplificación y secuenciación, así como procesos de fermentación y esterilización industrial.
Ver más56 páginas


Visualiza gratis el PDF completo
Regístrate para acceder al documento completo y transformarlo con la IA.
Vista previa
Tecnología del DNA Recombinante "in Vitro"
Ingeniería genética: se encarga de la manipulación/modificación "in vitro" de genes y la transferencia de la nueva molécula al organismo original u otro hospedador (basado en la universalidad del DNA) con un propósito determinado. Requiere conocer la secuencia de nucleótidos del gen/genes a manipular. Para ello debemos conocer una serie de métodos o técnicas para manipular el DNA.
Obtención del DNA
- Amplificación del DNA
- Electroforesis del DNA en gel
1 Secuenciacion de DNA
- Utilización de enzimas: nucleasas de restricción y ligasas
Hibridación de los ácidos nucleicos.
Métodos de Manipulación de DNA
1. Obtención del DNA
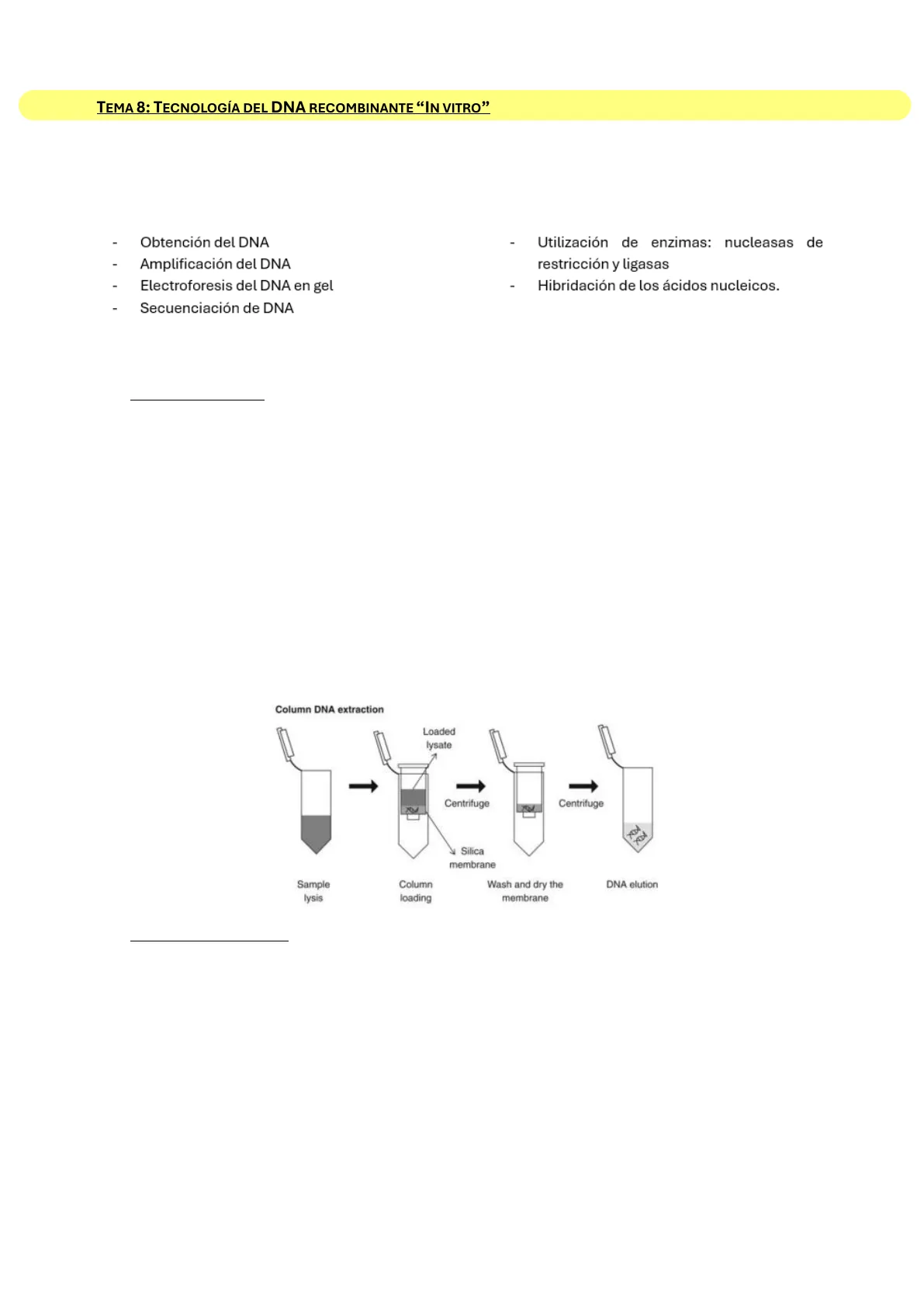
Para extraer el DNA que se encuentra en el núcleo existen muchos métodos. Lo que hacemos es romper todas las envolturas que hay alrededor del DNA. Para ello se tienen diferentes protocolos de actuación o kits de actuación. Estos se adaptan a la muestra y al grado de purificación. Los principales pasos que siguen son:
a) Ruptura de envolturas celulares (lisis celular): las membranas que protegen al DNA se rompen para liberarlo al medio. b) Eliminación de proteínas. La mayoría de las envolturas tienen una gran cantidad de proteínas. c) Lavado y recuperación de DNA.
Casi todas las extracciones se realizan con kits comerciales, pues son más limpios. La mayoría se basan en la utilización de columnas con filtros, estas tienen una membrana a la que une el DNA durante el proceso de obtención. En la columna se introducen también reactivos que limpian el DNA. Pasa por centrifugación por cada reactivo que se añade. Se tiene la columna en un tubo limpio y se añade un buffer.
Column DNA extraction Loaded lysate Centrifuge Centrifuge Silica membrane Sample lysis Column loading Wash and dry the membrane DNA elution
2. Amplificación del DNA
La amplificación de DNA se realiza mediante PCR, nos permite hacer muchas copias de un fragmento de DNA. Para llevarse a cabo es necesario una serie de elementos: primeramente, el DNA molde obtenido de la célula del estudio, enzimas polimerasas, oligonucleótidos complementarios al fragmento que queremos amplificar y nucleótidos. Los pasos a seguir son los siguientes:
a) Desnaturalización: mediante la aplicación de altas temperaturas se rompen los enlaces de hidrógeno que unen a las dos hebras del DNA, separándolas. b) Anillamiento: la temperatura que se aplique depende de la secuencia de los primers. c) Extensión: la polimerasa introduce los nucleótidos que corresponden para hacer la copia.
41La repetición del ciclo se realiza unas 35 veces a través de un termociclador. Se consigue un aumento exponencial del número de copias. La amplificación de la secuencia del DNA es necesaria para poder manipularlo y secuenciarlo.
3. Electroforesis del DNA en gel
La electroforesis sirve para separar moléculas de DNA en función de su carga y tamaño en un campo eléctrico. Normalmente se hace dentro de un gel de agarosa y utilizando una disolución tampón que es conductora. Las moléculas se desplazan a través de los poros del gel hacia el polo opuesto a su carga neta, es decir al polo positivo. La separación se produce rápido porque a menor tamaño de las moléculas de DNA mayor es la velocidad. Una vez separado el DNA, para verlo tiene que estar unido a una molécula que se intercala entre la doble hélice y que emita una señal (normalmente fluorescente), la molécula comúnmente utilizada es el bromuro de etidio. Aunque la adición de moléculas externas puede causar una mutación, como se ha estudiado anteriormente.
Para conocer el tamaño de las bandas obtenidas, se tiene que añadir un marcador de pesos moleculares. Estos son mezclas de moléculas de DNA comerciales que nos dan información sobre el tamaño de la molécula y sobre la cantidad de DNA. Si brilla, significa que hemos extraído DNA, cuanto más brilla, más cantidad de DNA.
4. Secuenciación de DNA
La manipulación de DNA se basa en conocer la secuencia de DNA con la que se quiere trabajar. La secuenciación del DNA es un proceso que nos permite conocer la secuencia nucleotídica de una molécula de DNA.
El procedimiento más conocido es la secuenciación de Sanger. Este proceso requiere la amplificación del segmento que se quiere secuenciar. El procedimiento que sigue es introducir el DNA molde en 4 tubos de ensayos diferentes a los que se les realiza una PCR a cada uno de ellos. Añadiendo didesoxinucleótidos se paraliza la amplificación, obteniendo cadenas de diversos tamaños acabadas todas en el nucleótido añadido.
En el estudio de los métodos de secuenciación se diferencian varias generaciones:
- Primera generación: se incluye la secuenciación de Sanger. Se trata de una secuenciación automatizada del DNA desarrollada en los años 90. Es un método menos nocivo que la fluorescencia y los radiactivos. Tiene una capacidad de lectura de 500-1000 bases. Se obtiene por ordenador el cronograma y la secuencia. Requiere como se ha comentado con anterioridad de dos PCR.
- Segunda generación: comienza con la secuenciación masiva. Inmovilización de reacciones en superficies sólidas. Se obtienen un mayor número de secuencias en un menor tiempo, empleando un menos número de reactivos y por ello supone un menor coste. Dependiendo del tipo de secuenciador, lo primero que se hace es fragmentar el DNA, se añaden adaptadores en el borde que inmovilizan. Las placas se llenan de trocitos de secuencias de DNA, después se añade de manera artificial un fragmento del complementario. Se produce una PCR en la que se hacen copias del fragmento a secuenciar y se graban los colores de los nucleótidos.
- Tercera generación: se emplean equipos tan sensibles que no son necesarias muchas copias de un segmento, con una única molécula es suficiente. A la molécula se le adhiere una polimerasa, pasan los reactivos y la nucleasa añade los nucleótidos por complementariedad. Se libera un polón. La ventaja de este avance es que se prescinde de la amplificación por PCR, y consiguiente una mayor unidad de secuencias por unidad de espacio en un menor tiempo, empleando una menor cantidad de reactivos y reduciendo costes.
5. Utilización de enzimas: nucleasas de restricción y ligasas
Enzimas de restricción: rompen los enlaces fosfodiéster que hay entre los nucleótidos de los ácidos nucleicos. Cada vez que se encuentra la secuencia determinada, la enzima la detecta y la rompe. Los cortes pueden ser de dos tipos: romos (perpendicular a la cadena, quedando dos cadenas cortadas por el mismo nucleótido) o cohesivos/escalonados (cada cadena es cortada por un nucleótido diferente). Los productos pueden ser unidos de nuevo mediante ligasas (vuelven a generar los enlaces de unión).
Las ligasas son enzimas que unen los extremos de DNA creando enlaces fosfodiester entre el extremo 5' de una molécula y el 3' de otra. Las ligasas forman parte de los procesos de replicación y recuperación natural de las células. En tecnología del DNA recombinante se utiliza DNA-ligasa T4 (ligasa del bacteriófago T4).
5-₽ 3'-OH 3-OH 5'-p. T4 DNA ligase T4 DNA ligase + ATP ATP MgCl MgCl 5'-P x-OH J'-OH 5-P ! Ligación de extremos cohesivos Sticky end DNA ligation Blunt end DNA ligation Ligación de extremos romos
6. Hibridación de los ácidos nucleicos
Para encontrar secuencias relacionadas en genomas diferentes o determinar si un gen se expresa (RNA) se usan dos técnicas:
- Técnica Southern: se hibridan sondas de secuencia conocida de DNA o RNA con fragmentos de DNA diana que se han separado por electroforesis en gel.
- Técnica Northern: se hibridan sondas de secuencia conocida de DNA o RNA con fragmentos de RNA diana que se han separado por electroforesis en gel.
Los fragmentos de DNA/RNA son desnaturalizados, se someten a electroforesis y se transmiten a una membrana. Se expone la membrana a una sonda de DNA/RNA marcada. Si la sonda es complementaria de alguno de los fragmentos se forman híbridos. La hibridación se puede detectar monitorizando la sonda marcada que se ha unido a la membrana.
Clonación Génica
Clonar: obtener copias iguales de un fragmento de DNA de interés (no necesariamente de DNA). Actualmente se extiende el concepto a la inserción del fragmento en un vector para expresar ese segmento en otro organismo.
Fases de la clonación molecular:
- Amplificación por PCR del fragmento de DNA a clonar. Necesitamos muchas copiar.
- Digestión con enzima de restricción del fragmento a clonar y del vector de clonación. Recortamos por las esquinas para quedarnos solo con la secuencia sencilla.
- Inserción del fragmento en el vector mediante DNA ligasa.
- Inserción del vector de clonación en el hospedador.
435. Seleccionar los hospedadores que contienen el vector de clonación, es decir, los recombinantes. Tienen un marcador de selección para diferenciar las células que han adquirido la construcción que hemos realizado.
Vectores de clonación: son moléculas de ácido nucleico (DNA) que nos sirve para incorporar fragmentos de DNA exógeno en un hospedador. Debe de cumplir algunas propiedades:
- Capacidad de replicarse en el hospedador. Es necesario que al dividirse se replique también en el nuevo organismo pues en el caso contrario el fragmento insertado desaparecerá en las células hijas.
- Región múltiple de clonación, MCS: zona en la que hay muchísimas secuencias reconocidas por enzimas de restricción.
- Marcador de selección: gen que permite seleccionar cuales de los fragmentos han recibido el vector.
- Fáciles de introducir/recuperar en/de la célula huésped.
Secuencia codificante del gen heterólogo MCS RBS Promotor Terminador Casete de expresión Origen de replicación Marcador de selección Figura 9.12. Elementos principales de un vector de ex- presión autoreplicativo. RBS (ribosome binding site); MCS (multiple cloning site).
Se distinguen diferentes tipos dependiendo de su aplicación: depende del tamaño máximo a clonar, del marcador de selección, de los sitios de restricción, número de copias que puede mantener la célula hospedadora. Son también importantes otros elementos genéticos para controlar la expresión del gen clonado (promotores) ...
Plásmidos
Los plasmidos son moleculas de DNA de doble cadena extracromosómicas, generalmente circulares, aparecen en bacterias, levaduras ... Se dividen de forma independiente. Pueden portar genes para la selección (como genes de resistencia a antibióticos). Permiten clonar fragmentos pequeños. No todos pueden replicarse y mantenerse, depende del hospedador algunos podrán replicarse y otros no.
Selectable Marker Promoter 5'Primer Site Restriction Site Plasmid Map Inserted Gene Restriction Site 3' Primer Site Antibiotic Resistance Gene Los plásmidos permiten clonar fragmentos cortos de DNA de hasta 15Kb. Sin embargo, no todos los plásmidos pueden replicarse y Origin of Replication mantenerse en todas las especies. Los plasmidos sirven para la selección de bacterias transformadas por resistencias a los antibióticos.
Ejemplo: contiene el gen lacZ (enzima ß-galactosidasa) con el MCS.
Fagos y Cósmidos
Los fagos son virus bacterianos que permiten clonar mas cantidad de bases. Introducen su material genético por transducción.
Los cósmidos son vectores híbridos obtenidos a partir del fago lambda y DNA plasmídico. Codifican para la cubierta del fago. Permiten que el DNA si se empaquete y fago se pueda introducir dentro de la bacteria por transducción.
El fago más conocido es el fago lambda, es un bacteriófago de DNA de cadena lineal doble.
Existen muchas variantes de fagos: según las regiones de clonación, los sistemas de detección ... También se puede controlar la integración en el genoma o el desencadenamiento del ciclo lítico.
44
¿Non has encontrado lo que buscabas?
Explora otros temas en la Algor library o crea directamente tus materiales con la IA.
