Farmacocinética y fármacos cardiovasculares: absorción, distribución y eliminación
Esquemas de Farmacocinética sobre farmacocinética, incluyendo absorción, distribución, metabolismo y eliminación de fármacos. Los resúmenes detallan vías de administración, influencia del pH y tablas comparativas de fármacos como Fosinopril y Losartán, útiles para estudiantes universitarios de Biología.
Ver más26 páginas


Visualiza gratis el PDF completo
Regístrate para acceder al documento completo y transformarlo con la IA.
Vista previa
Farmacocinética
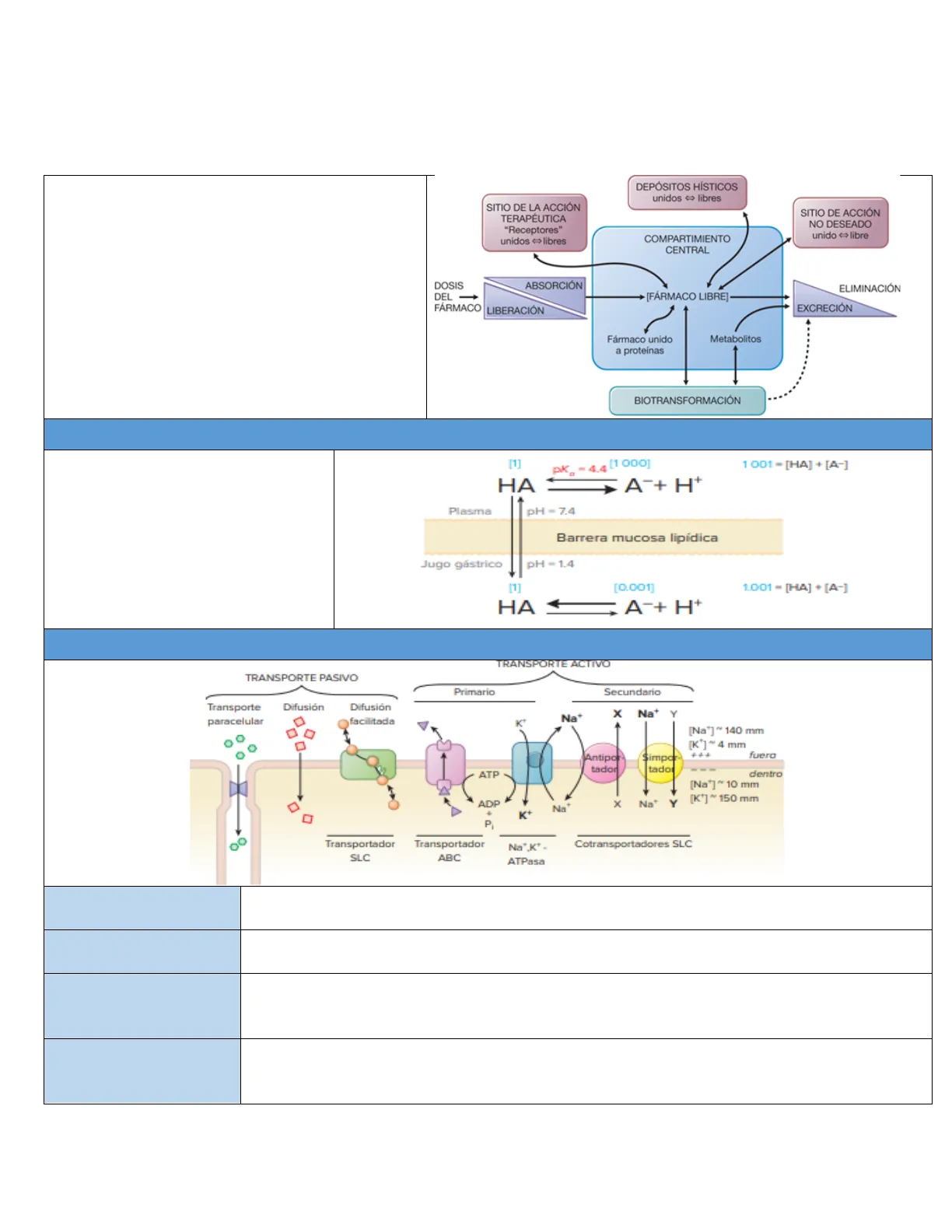
Interacción y movimiento de los fármacos en el cuerpo (Interacción del cuerpo con el fármaco) desde su administración, hasta su eliminación por medio del proceso ADME
Adsorción
- Distribución
- Metabolismo
- Eliminación
DEPÓSITOS HÍSTICOS unidos > libres
SITIO DE LA ACCIÓN TERAPÉUTICA "Receptores" unidos >libres
SITIO DE ACCIÓN NO DESEADO unido >libre
COMPARTIMIENTO CENTRAL
ABSORCIÓN ELIMINACIÓN + [FÁRMACO LIBRE] EXCRECIÓN LIBERACIÓN
Fármaco unido a proteínas
Metabolitos
Biotransformación
Influencia del pH en la distribución
Influencia del pH en la distribución de un ácido débil (pKa = 4.4) entre el plasma y el jugo gástrico separados por una barrera lipídica
PH [1] PK_ - 4.4 [1 000] 1 001 - [HA] + [A-] HA A + H+
Plasma PH -7.4
Barrera mucosa lipidica
Jugo gastrico PH -1.4 [1] [0.001] 1.001 - [HA] + [A-] HA - A-+ H+
Transporte de membrana mediado por transportador
Transporte activo
Primario Secundario
Transporte pasivo
Transporte paracelular
Difusión Difusión facilitada
Na+ K+ [Na]]~140 mm [K]~ 4 mm +++ fuera
Antipor tador
Simpor tador
dentro [Na]~10 mm ADP x Na+ Y + K+ P
Transportador SLC
Transportador ABC
NaT.K. ATPasa
Difusión pasiva
Movimiento impulsado por gradientes de concentración. Difusión simple desde áreas de alta concentración a áreas de baja concentración sin gasto de energía.
Difusión facilitada
Moléculas atravesadas con la ayuda de transportadores o proteínas facilitadoras. Ocurre a favor de un gradiente de concentración sin consumo de energía.
Transporte activo
Paso de sustancias facilitado por transportadores en la membrana celular. Independiente del gradiente de concentración, incluso en contra de este. Energía proporcionada por la hidrólisis del ATP.
Pinocitosis
Englobamiento de partículas o líquidos por la célula. Permite la entrada de macromoléculas. Involucra la invaginación de la membrana celular, formando vesículas que se liberan en el citoplasma.
x 4 Na+ Y ATP [K]~ 150 mm Na
Cotransportadores SLC
Vías de administración
DOSIS DEL -+ FÁRMACOVÍAS DE ADMINISTRACIÓN
Enterales
Oral: Administración a través de la boca. Sublingual: Colocación bajo la lengua. Rectal: Introducción en el recto
Parenterales
Intradérmica: inyección en la dermis. Intravenosa: administración en la vena. Intramuscular: inyección en el músculo. subcutánea: administración bajo la piel.
Otras vías
OFTÁLMICA ÓTICA NASAL VAGINAL INTRAOCULAR INHALATORIA TÓPICA TRANSDÉRMICA
Drug concentration in blood (c) Intravenous Intramuscular Subcutaneous Oral Time (t)
Características de las vías comunes de administración de fármacos
TABLA 2-1 Algunas características de las vías comunes de administración de fármacosa
VÍA Y BIODISPONIBILIDAD (F)
PATRÓN DE ABSORCIÓN
UTILIDAD ESPECIAL
LIMITACIONES Y PRECAUCIONES
Intravenosa F - 1 por definición
Evita la absorción
Valiosa para uso de emergencia Mayor riesgo de efectos adversos
Efectos potencialmente inmediatos
Permite la titulación de la dosis Como regla deben inyectarse las solucio- nes lentamente
Adecuado para grandes volume- nes y para sustancias irritantes, o mezclas complejas, cuando se diluye
Por lo general, se requiere para proteí- nas de alto peso molecular y fármacos péptidos
No es adecuado para soluciones oleosas o sustancias poco hidrosolubles
Subcutánea 0.75 < F < 1
Rápido en solución fase acuosa
Adecuado para algunas suspensiones poco solubles y para la instilación de implantes de liberación lenta
No es adecuado para grandes volúmenes
Lento y sostenido de los prepara- dos de depósito
Posible dolor o necrosis por sustancias irritantes
Intramuscular 0.75 < F < 1
Rápido en solución fase acuosa
Adecuado para volúmenes moderados, vehículos aceitosos y algunas sustancias irritantes
Evitar durante la terapia anticoagulante
Lento y sostenido de los prepara- dos de depósito
Apropiado para la autoadministración (p. ej., insulina)
Puede interferir con la interpretación de ciertas pruebas de diagnóstico (p. ej., creatina cinasa)
Ingestión oral 0.05< F < 1
Variable, depende de muchos factores (véase texto)
Más conveniente y económico; general- mente más seguro
Requiere conformidad del paciente Biodisponibilidad potencialmente errá- tica e incompleta
Liberación
LIBERACIÓN La liberación de un principio activo de una forma farmaceutica es crucial para su eficacia y seguridad.
Bioequivalencia
son fármacos los cuales poseen una misma eficacia y seguridad
Absorción
desplazamiento de un fármaco desde su lugar de administración hacia el compartimiento central
Biodisponibilidad
grado fraccionario en que una dosis administrada del fármaco alcanza su sitio de acción
F = Cantidad de farmaco que alcanza la circulación sistémica Cantidad de farmaco administrado
Distribución
DISTRIBUCIÓN Paso de un fármaco desde la circulación general a los líquidos intersticiales e intracelulares
Factores determinantes: GC, Flujo sanguíneo regional, Permeabilidad capilar, Volumen hístico Factores que afectan: liposolubilidad, unión a proteínas plasmáticas y los gradientes de pH
Transporte en la sangre
Los fármacos pueden viajar libremente en el plasma, unirse a proteínas plasmáticas (como la albúmina) o incorporarse en células, principalmente eritrocitos. La unión a proteínas plasmáticas facilita el transporte del fármaco en la sangre hacia diferentes tejidos.
Depósitos tisulares y redistribución
Los fármacos se pueden concentrar en tejidos en mayor cantidad que en el plasma, y esto no siempre corresponde al órgano o tejido diana. Los depósitos tisulares incluyen grasa neutra (fármacos lipofílicos), hueso, hígado, pulmón y piel.
Barreras
Barrera Hematoencefálica: Estructura limitante que regula el paso de sustancias hacia el tejido intersticial nervioso. Barrera Placentaria: Regula el paso de sustancias entre la circulación sanguínea fetal y materna durante el embarazo.
Distribución del flujo sanguíneo en varones
TABLA 2-2 Distribución del flujo sanguíneo en varones de 70 kg de peso en reposo
RIÑONES CORAZÓN HÍGADO CEREBRO MÚSCULOS ESQUELÉTICOS GRASA RESIDUO Σ
Flujo sanguíneo (mL/min) 1 100 250 1700 800 900 250 500 5500
Masa (kg) 0.3 0.3 2.6 1.3 34 10 21.5 70
Flujo/masa (mL/min/kg) 3 667 833 654 615 26 25 23
% de gasto cardiaco 20 4.5 31 14.5 16.4 4.5 9.1 100
70 - 60 - Sangre y órganos magros (corazón, cerebro, hígado, pulmón y riñones)
Compartimiento hístico Musculo, piel, grasa y hueso
Sangre Adiposo Músculos esqueléticos Hígado
Farmaco eliminado
10 - 0 - 0 5 10 15 20 30 60 90 120 Tiempo, minutos
IV dosis
Compartimiento central
Concentración de tiopental, unidades arbitrarias
- 20 min 90 min
20 -
Fases de distribución
Fase inicial Órganos de alto flujo sanguíneo: hígado, riñones, encéfalo · Rápida captación inicial
Segunda fase Tejidos con mejor irrigación: músculos, piel y grasa · Equilibrio más lento (minutos a horas).
Unión a proteínas plasmáticas
Efectos: . Reduce concentración libre activa . Limita filtración glomerular. . Modifica transporte y metabolismo
T 0+
Redistribución
Movimiento del fármaco desde el sitio de acción a otros tejidos Ejemplo (Tiopental: rápida acción anestésica seguida de redistribución a músculos)
Metabolismo
Transformación de fármacos lipofílicos en metabolitos hidrofílicos facilita la excreción renal y termina la actividad biológica
Factores que afectan: Variaciones genéticas Inducción/inhibición enzimática · Por fármacos · Enfermedades y factores étnicos/genero. . Impactan la actividad de las enzimas (farmacogenética).
Cinética del primer orden
La cantidad de fármaco metabolizado por unidad de tiempo es proporcional a la concentración plasmática del fármaco (la fracción del fármaco eliminado por el metabolismo es constante)). Eliminación proporcional a la concentración plasmática
CONCENTRACIÓN PLASMÁTICA C 32 Cp = 31 16 DEL FÁRMACO 8 (ug/ml) 4 2 t1/2 1 2 4 6 8 10 12 TIEMPO (h)
Cinética de orden cero
Se metaboliza una cantidad constante de fármaco por unidad de tiempo / Eliminación fija, ocurre con saturación
CONCENTRACIÓN PLASMÁTICA 32 CP 16 V = Dosis /Cp DEL FÁRMACO 8 (ug/ml) 4 2 t1/2 1 O 2 4 6 8 1℃ 12 TIEMPO (h)
Enzimas biotransformadoras inducidas
Metabolito producto resultante del metabolismo
Profármaco
compuestos inactivos que se convierten a sus formas activas por medio del metabolismo Ventajas: Incremento de la biodisponibilidad · Activación en el sitio de acción Ejemplos: Enalapril: convertido en enalaprilat por hidrólisis · Clopidogrel: activado por CYP450 para inhibir la agregación plaquetaria
Biotransformación
proceso por el cual se modifica la estructura química de un fármaco para que sea más fácil su eliminación
La biotransformación implica:
Fase 1
Incluye oxidación, oxigenación, reducción, hidrólisis, alquilación, desalogenación. Realizado por enzimas catabólicas en la fracción microsomal de células. Modificaciones basadas en la obtención de grupos funcionales.
Fase 2
Sustancias generadas en la Fase 1 se unen covalentemente a sustancias endógenas como ácido glucurónico Requiere la participación de transferasas que catalizan la conjugación de xenobióticos con moléculas mencionadas.
Citocromo P450
Definición: Complejo multienzimático del sistema de las monooxigenasas. Importancia: Aproximadamente la mitad de los fármacos son metabolizados por este citocromo. Distribución: Presente en varios tejidos (pulmón, riñón, piel, intestino, placenta), especialmente activo en el hígado. Sensibilidad: Muy sensible a la inducción por fármacos.
Excreción
EXCRESIÓN Después de pasar por los procesos metabólicos, el último paso para un fármaco es la excreción, eliminando tanto la sustancia como sus metabolitos del organismo.
Principales vías de excreción
Renal: incluye filtración glomerula, secreción activa y reabsorción pasiva. Biliar y fecal: excreción a través de la bilis y heces. Pulmonar: eliminación de gases anestésicos. Leche materna: fármacos lipofílicos y alcalinos pueden concentrarse en la leche
Excreción renal
Eliminación principal de fármacos inalterados o como metabolitos activos mediante la utilización del sistema renal Recibe hasta el 25% del gasto cardíaco.
Arteriolas aferente eferente
Glomérulo
Fármaco activo
Túbulo proximal
Secreción
Red /capilar peritubular -
Asa de Henle
Reabsorción tubular
Reabsorción Tubular Fármacos filtrados pueden ser reabsorbidos por células tubulares y retornar a la circulación. Principalmente ocurre en túbulos distales y proximales por difusión simple.
Túbulo distal
Fármaco Reabsorción + Orina
Parámetros que rigen la disposición de los fármacos
Los siguientes son los cuatro parámetros más importantes que rigen la disposición de los fármacos:
Biodisponibilidad definida como la fracción del farmaco absorbido como tal en la circulación sistémica.
Volumen de distribución una medida del espacio aparente en el cuerpo disponible para contener el fármaco en función de cuánto se administra contra lo que se encuentra en la circulación sistémica.
Aclaramiento o depuración una medida de la eficiencia del cuerpo para eliminar el fármaco de la circulación sistémica.
Tiempo de vida media de eliminación (t1/2) una medida de la velocidad de eliminación del fármaco de la circulación sistémica.
Cálculo de dosis mantener la concentración plasmática en estado de equilibrio para eficacia terapéutica.
Filtración glomerular
Filtración Glomerular Ocurre en los capilares del glomérulo, donde poros permiten el paso de sustancias. Fármacos no unidos a proteínas pueden ser filtrados. Factores como la edad y enfermedades renales afectan la cantidad de fármacos excretados.
Secreción tubular
Secreción Tubular Células de los túbulos renales excretan fármacos al lumen tubular. Transporte activo regulado por diversos factores.
¿Non has encontrado lo que buscabas?
Explora otros temas en la Algor library o crea directamente tus materiales con la IA.
