Eliminazione dei farmaci e farmacologia pediatrica: meccanismi e fattori
Slide di Carlotta Fedrizzi Maria Rosaria Sannino Pharmacology – Salvatorelli Emanuela su Drug elimination. Il Pdf esplora i meccanismi di eliminazione dei farmaci, differenziando tra cinetica di ordine zero e di primo ordine, con un focus sulla farmacologia pediatrica e il metabolismo dei farmaci, utile per studenti universitari di Biologia.
Mostra di più18 pagine


Visualizza gratis il Pdf completo
Registrati per accedere all’intero documento e trasformarlo con l’AI.
Anteprima
Carlotta Fedrizzi
Maria Rosaria Sannino
Pharmacology - Salvatorelli Emanuela
25.03.2024
Drug elimination
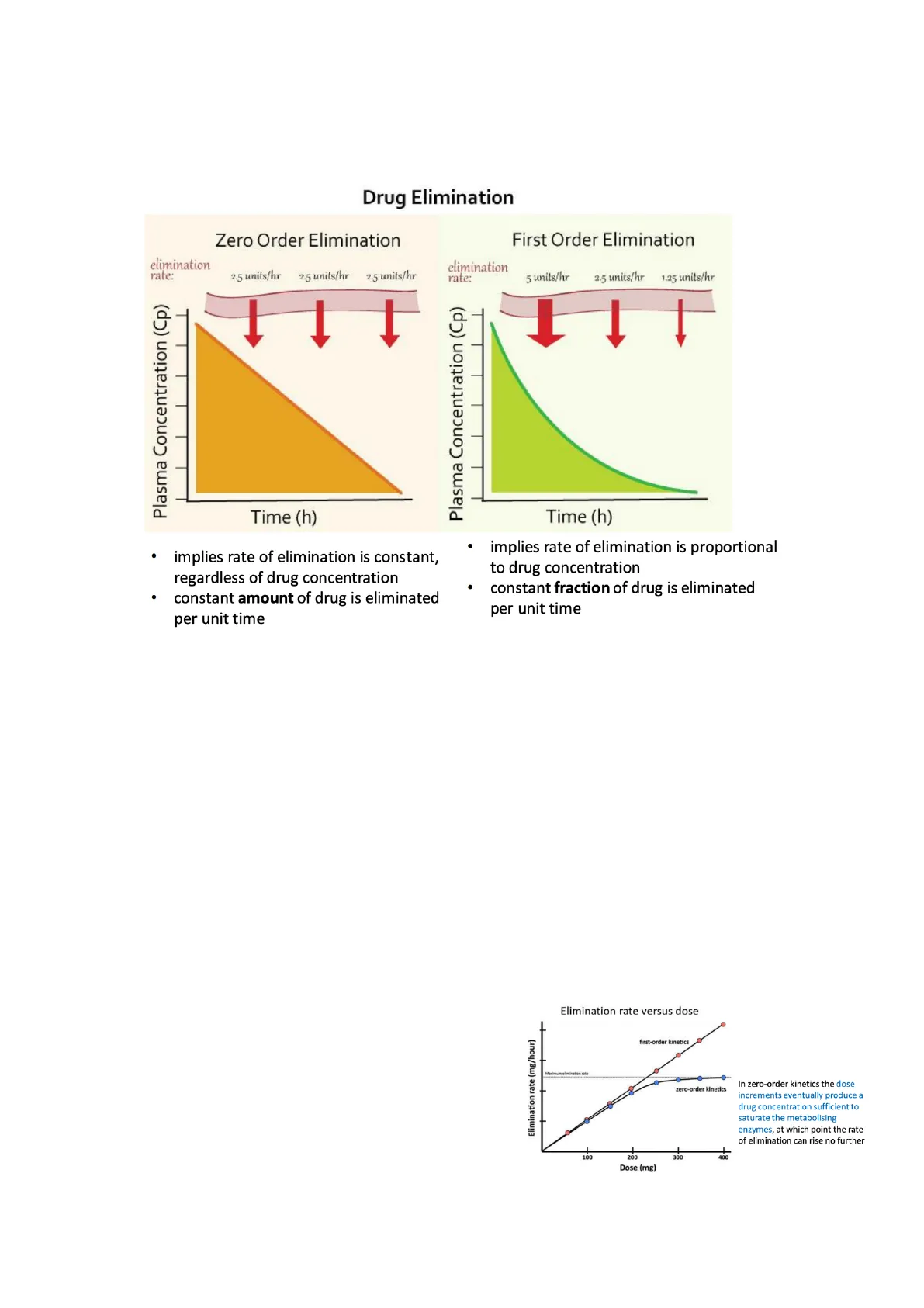
Drug Elimination
Zero Order Elimination
elimination rate: 2.5 units/hr 2.5 units/hr 2.5 units/hr
- implies rate of elimination is constant, regardless of drug concentration
- constant amount of drug is eliminated per unit time
First Order Elimination
elimination rate: 5 units/hr 2.5 units/hr 1.25 units/hr
- implies rate of elimination is proportional to drug concentration
- constant fraction of drug is eliminated per unit time
Drugs are generally eliminated following a first order elimination, only some drugs are eliminated following a kinetic of zero order elimination. Drugs eliminated following first order kinetic, in this case the elimination is directly proportional to the concentration of the drug, the rate of elimination is proportional to drug concentration. (rate of elimination means the velocity of elimination of an amount of drug concentration). Velocity of elimination is not constant, and we obtain a curve (on the right). Zero order kinetic elimination is different, because in this case the velocity of elimination is constant, regardless of drug concentration, so we obtain this kind of curve (on the left). First order kinetic elimination is better because velocity is not constant, but it is proportional to the amount of drug concentration. In zero order case, what could happen to the drug? If I have a lot of concentration, you can have toxicity, because the drug could be accumulated. e.g. alcohol eliminated by zero order kinetics, because the velocity of alcohol elimination is constant, so in the case of zero order kinetics, the dose increments eventually produce a drug concentration sufficient to saturate the metabolising enzymes. The root of elimination of a drug that is eliminated by zero order kinetics could be saturated, so we are not able to eliminate the drug, so it accumulates.
Elimination rate versus dose
In zero-order kinetics the dose increments eventually produce a drug concentration sufficient to saturate the metabolising enzymes, at which point the rate of elimination can rise no further
Effect of saturation on maintenance of steady state
Zero order kinetics: Process required for elimination is saturated. For some drugs, elimination processes can be saturated (i.e. when metabolism is required). At sufficiently high concentrations, when the dose exceeds that which can be eliminated, regular dosing no longer produces a steady state. The increasing of the Css will be out of proportion Css= concentration at the steady state; it is a concentration in the plasma for a drug in which the concentration that enters in the plasma is equal to the concentration that is eliminated from the plasma. The maintaining dose is important to reach a steady state of a drug, especially for administration of a drug for a long time. So, to reach the concentration at the steady state is important because it means that the concentration of the drug is optimal for inducing a response. For drugs eliminated with zero order kinetics, it's very difficult to achieve the Css, because the drug tends to accumulate; on the other hand, for a drug eliminated with first order kinetic it is not a problem, the Css can be easily reached. A drug that follows a zero order kinetic, it is more difficult to manage, because its pharmacokinetic is not linear in relation to the concentration of the drug.
Metabolism
ADME:
- absorption,
- distribution,
- metabolism
- elimination
Pharmacokinetics
The principles o ADME
Absorption
How will it get in?
Distribution
Where will it go?
Metabolism
How is it broken down?
Elimination
How does it leave?
For the metabolism of a drug, we don't have a parameter. What is the metabolism of a drug? Biotransformation. When is it important to transform a drug? If we administer a drug with a great lipophilic characteristic, what drug is better absorbed, a lipophilic or hydrophilic? Lipophilic, whereas which one is better eliminated? Hydrophilic. Therefore how can we eliminate a lipophilic drug? Biotransforming/ metabolising the drug, in the sense that we have to transform the parent drug in more polar metabolites.
PHARMACOKINETIC PARAMETERS
Absorption
Movement of drug from site of administration into circulation
Distribution
Pattern of scatter of specified amount of drug among various locations in the body
Metabolism
Enzyme catalysed chemical transformation of drugs within living organism
Elimination
Excretion of drug metabolites outside the body
Drug metabolism
A drug can undergo a metabolism to obtain more polar metabolites, and to facilitate it's excretion. E.g. a lot of benzodiazepines have a long half-life, because they are very lipophilic, so they are intensely metabolised. What is biotransformation? It's a chemical alteration of the drug in the body. If we have a hydrophilic drug, this kind of drug is easily eliminated by the renal elimination system, so easily excreted.
The Effect of Drug Metabolism on Excretion
Lipophilic (or fat-soluble) drugs are metabolized to form relatively more hydrophilic (or water-soluble) metabolites than the parent drug, and these metabolites are thus more easily excreted. To convert non-polar compounds to polar compounds to avoid reabsorption in renal tubules If we have a lipophilic drug, if it arrives in the glomeruli, probably this drug is retained/ reabsorbed, as it's not polar enough to be easily excreted. So, this lipophilic drug is easily delivered to the liver, where it is transformed to more polar metabolites, that are easily eliminated.
The liver is the organ that most easily transforms a lipophilic drug, but there are other organs that are able to do this: kidneys, lungs, adrenal glands, placenta, brain, skin, plasma.The liver is the most important organ able to metabolise a drug, because it has a lot of metabolising enzymes, they can be microsomal or non-microsomal,
- microsomal enzymes are in smooth ER of the liver, e.g. CYP-450, or monooxygenase etc.
- non-microsomal enzymes are localised in the cytoplasm and in the mitochondria, e.g. alcohol dehydrogenase or MAO (mono amino oxidase)
Process of metabolism is usually divided in two phases, first phase 1 reaction, then phase 2 reaction
Phase 1 reaction
Types of reactions: Reduction, oxidation and hydrolysis. These reactions introduce functional chemical groups that are polar chemical groups, the most important is the oxidation. Oxidation reactions are mediated by cytochrome P 450 (CYP450) enzymes in the liver; there are a lot of isoforms of this enzyme: look at pie chart on the below on the left.
Metabolism is often divided into two phases of biochemical reaction
Phase 1 Reaction
- It is called Functionalization Reaction.
- Function: Introduction of functional groups such as -OH, -NH2, -SH, -COOH into the compound to produce more water soluble compound.
- Reaction type: Oxidation, Reduction and Hydrolysis.
Phase 2 Reaction
- It i is called Conjugation Reaction.
- Function: Conjugation of a compound or its metabolites with endogenous substrate to form water soluble conjugated products.
- Reaction type: Glucuronidation, Glutathione sulfation, conjugation Acetylation and Methyalation.
Role of CYP Enzymes in Hepatic Drug Metabolism
CYP 3A4 and CYP3A5 are the most abundant in the liver cells, they are 26% of the relative hepatic content. If you look at pie chart on the right of the image, CYP 3A4 and CYP3A5 are able to oxidise 33% of the total of the drug, so these two isoforms are the most abundant and the most active.
CYP450 catalytic cycle
Cycle that is able to oxidise a drug. In this cycle we have the drug in its lipophilic form, the CYP450 that is bound to the heme rings, then the NADPH and the oxygen are important to obtain an oxidised drug form. These kinds of enzymes are also called mixed function oxidases (enzymes). Generally, drugs can be metabolised after phase 1 reaction, or after phase 2 reaction; majority of drugs are metabolised after phase 1 and then phase 2 sequentially, sometimes some drugs can be metabolised by phase 1 and 2 but not sequentially. If a drug undergoes phase 1 reaction, and we obtain a sufficiently hydrophilic product to be eliminated, if not it can undergo phase 2 reactions.
Phase 2 reactions
They are conjugation reactions. Their aim is to conjugate a compound or a metabolite with endogenous substrate (polar) to form water soluble conjugated products, that are very polar and easily eliminated. What enzymes are able to mediate this reaction? Transferases, found mainly in the liver but also in other tissues and organs, e.g. intestine.
Phase 1 Reaction
- It is called Functionalization Reaction.
- Function: Introduction of functional groups such as -OH, -NH2, -SH, -COOH into the compound to produce more water soluble compound.
- Reaction type: Oxidation, Reduction and Hydrolysis.
Phase 2 Reaction
- It is called Conjugation Reaction.
- Function: Conjugation of functional groups of a compound or its metabolites with endogenous substrate to form water soluble conjugated products.
- Reaction type: Glucuronidation, sulfation, Glutathione conjugation Acetylation and Methyalation.
The products may not be sufficiently hydrophilic but are suitable precursors for phase II/ Conjugation. They are called transferases because they transfer an endogenous compound to the metabolite of phase 1 reaction or to compound. The most important and most common phase 2 reaction is glucuronidation. Phase 1 -> phase 2 -> transferases -> glucuronic acid (endogenous compound). Glucuronidation includes the transfer of glucuronic acid to the molecule. UGT are these specific enzymes that transfer the glucuronic acid stands uridine 5'-diphospho- glucuronosyltransferase. Phase II reactions typically involve conjugation with glucuronic acid, as endogenous substrate. This makes the drug water-soluble and suitable for renal excretion.
Other phase 2 reactions
Glucuronidation
Glucoronyl Transferase -OH,-COOH, -NH2, -SO2NH2
Sulfation
Sulfotransferase -OH, -NH2, -SO2NH2, -NHOH
Glutathione
Glutathione-S-transferase Alkyl Halides, Alkyl Nitrates, Epoxides, Lactones (electrophilic centers)
Acetylation
Acetyl transferase -OH, -SO2NH2, -NHNH2, -NH2, -NHOH
Amino Acid Conjugation
Acyl transferase -COOH,
Methylation
Methyl transferase -OH, -NH2, -SH
Non hai trovato quello che cercavi?
Esplora altri argomenti nella Algor library o crea direttamente i tuoi materiali con l’AI.
